Article Text

Abstract
Background: Short stature affects approximately 2% of children, representing one of the more frequent disorders for which clinical attention is sought during childhood. Despite assumed genetic heterogeneity, mutations or deletions of the short stature homeobox-containing gene (SHOX) are found quite frequently in subjects with short stature. Haploinsufficiency of the SHOX gene causes short stature with highly variable clinical severity, ranging from isolated short stature without dysmorphic features to Léri-Weill syndrome, and with no functional copy of the SHOX gene, Langer syndrome.
Methods: To characterise the clinical and molecular spectrum of SHOX deficiency in childhood we assessed the association between genotype and phenotype in a large cohort of children of short stature from 14 countries.
Results: Screening of 1608 unrelated individuals with sporadic or familial short stature revealed SHOX mutations or deletions in 68 individuals (4.2%): complete deletions in 48 (70.6%), partial deletions in 4 (5.9%) and point mutations in 16 individuals (23.5%). Although mean height standard deviation score (SDS) was not different between participants of short stature with or without identified SHOX gene defects (–2.6 vs –2.6), detailed examination revealed that certain bone deformities and dysmorphic signs, such as short forearm and lower leg, cubitus valgus, Madelung deformity, high-arched palate and muscular hypertrophy, differed markedly between participants with or without SHOX gene defects (p<0.001). Phenotypic data were also compared for 33 children with Turner syndrome in whom haploinsufficiency of SHOX is thought to be responsible for the height deficit.
Conclusion: A phenotype scoring system was developed that could assist in identifying the most appropriate subjects for SHOX testing. This study offers a detailed genotype-phenotype analysis in a large cohort of children of short stature, and provides quantitative clinical guidelines for testing of the SHOX gene.
- BMI, body mass index
- ISS, idiopathic short stature
- LWS, Leri-Weill syndrome
- PCR, polymerase chain reaction
- SDS, standard deviation score
- SHOX, short-stature homeobox containing gene
- chondrocytes idiopathic short stature
- Langer syndrome Léri-Weill syndrome
- Turner syndrome.
Statistics from Altmetric.com
- BMI, body mass index
- ISS, idiopathic short stature
- LWS, Leri-Weill syndrome
- PCR, polymerase chain reaction
- SDS, standard deviation score
- SHOX, short-stature homeobox containing gene
Short stature is a developmental, multifactorial condition with a strong genetic component. Although several genes have been reported to be associated with longitudinal growth, they account only for a small proportion of cases of short stature.1,2 One such gene is the short-stature homeobox-containing gene (SHOX), which encodes a homeodomain transcription factor3 and is believed to be responsible for the height deficit associated with Turner syndrome.4–6 Mutations in the SHOX gene are one of the more frequent genetic causes of short stature in individuals with isolated or familial short stature.7,8,9,10,11
Significant evidence points to an important role for SHOX as a mediator of linear growth. First, the gene is expressed in the developing skeleton during fetal life6 and is specifically expressed in bone marrow fibroblasts and proliferating and hypertrophic chondrocytes3–5,12,13; second, deficiency of SHOX at the growth plate is associated with marked disorganisation of chondrocyte proliferation14; and third, there is a dose-dependent association between the number of active copies of the SHOX gene and height, such that SHOX haploinsufficiency is associated with short stature, whereas SHOX overdose as seen in sex-chromosome polyploidy is associated with tall stature.15
More than 50 different mutations in the SHOX gene associated with short stature in subjects previously described as “idiopathic” or in subjects with Léri-Weill syndrome (LWS) or Langer syndrome have been reported and assembled in the SHOX gene mutation database (www.shox.uni-hd.de).16 This database has been established to provide clinicians and scientists access to a central source of information about SHOX mutations. Indeed, the phenotype can vary from extreme dwarfism, mesomelia (short forearms and short lower legs) and limb deformity as seen in Langer syndrome (two defective or absent SHOX alleles) to disproportionate short stature and mesomelia as seen in LWS and Turner syndrome (one absent SHOX allele), to apparently isolated short stature with no other obvious clinical signs, often referred to as “idiopathic” short stature.5,17,18 The typical mesomelia and Madelung deformity (clinical finding of dinner fork-like deformity of the wrist) of LWS are more prevalent in females, but often do not become apparent before puberty.10,19,20 The clinical phenotype represents a continuum from very severe, disproportionate short stature to milder short stature with or without a number of other clinical and radiological abnormalities, making the distinction between LWS and non-LWS rather arbitrary. Even within a single family with an identical genetic defect in all affected members, the phenotype can vary considerably.21 This phenotypic variability is seen in many other genetic disorders, such that the classic boundaries between syndromic and non-syndromic forms are blurred, although identical mutations are present.22 Thus, there is no reliable basis for strictly distinguishing syndromic and non-syndromic phenotypes caused by the same mutation genotypes.
The aim of this study was to determine the phenotypical and molecular spectrum of SHOX deficiency in a large group of children with short stature and to develop a scoring system to aid in future selection of the most appropriate patients for diagnostic SHOX testing.
PARTICIPANTS AND METHODS
The study was conducted in accordance with the principles of the Declaration of Helsinki/Good Clinical Practice and was approved by the ethics committees of the participating institutions. Investigators obtained written, informed consent from the participants’ parent(s) or legal guardian(s) before conducting study-related procedures.
Participants
We studied 1608 pre-pubertal children with short stature from 14 countries: Belgium (n = 96), Canada (n = 96), Croatia (n = 74), Czech Republic (n = 171), France (n = 138), Germany (n = 200), Hungary (n = 55), India (n = 216), Netherlands (n = 6), Poland (n = 93), Russia (n = 23), Spain (n = 181), Turkey (n = 114), USA (n = 145). The participants’ ethnic origins were: Caucasian 1348 (83.8%), Western Asian 227 (14.1%), Eastern Asian 9 (0.6%), Hispanic 10 (0.6%), African–American 7 (0.4%) and others 7 (0.4%). Subjects were invited to participate in genetic screening for this study if they fulfilled the following entry criteria: chronological age ⩾3 years and pre-pubertal (males: genital stage Tanner 1 and testes ⩽2 ml; females: breast stage Tanner 1); height <3rd percentile of the local reference range or height <10th percentile with height velocity <25th percentile; bone age <10 years for boys and <8 years for girls or <9 years for patients with Turner syndrome; no GH deficiency or GH resistance; no chronic disease; and no known growth-influencing medications.
Study participants were screened for deletions or mutations in the SHOX gene to determine their eligibility for participation in a growth hormone intervention study23. This cohort of participants with short stature does not overlap with participants from a previous study that investigated the prevalence of SHOX deficiency in children with idiopathic short stature (ISS)7. For comparison, a group of participants with Turner syndrome (n = 33) was included. The ethnic backgrounds of these participants were: 24 Caucasian (72.7%), 7 Western Asian (21.2%) and 2 Eastern Asian (6.1%).
Clinical assessments
Height was measured with calibrated stadiometers, and a number of anthropometric measures were assessed using standardised procedures and equipment to quantify possible disproportion: sitting height, arm span, forearm and upper arm length, lower leg length and the circumferences of forearm, upper arm, lower leg and upper leg.24,25 Investigators were trained on these anthropometric techniques at study initiation. In addition, investigators were requested to report the presence or absence of a number of defined dysmorphic signs often observed in patients with LWS that were listed on the case report forms.
DNA analyses
DNA preparation and PCR
Genomic DNA was prepared from 0.5 ml peripheral blood using the PUREGENE DNA Isolation kit (Gentra Systems, Minneapolis, MN, USA). PCR were performed as previously described by Rappold et al, 2002.7
PCR-based microsatellite analysis
To look for evidence of a SHOX gene deletion, two microsatellites in the vicinity of the SHOX locus (CA-SHOX, CAII) were PCR-amplified and analysed on polyacrylamide gels for homo- or heterozygosity for the size of the microsatellite. Genomic DNA (100 ng) was amplified with 20 pmol of each of the primers DXYS201 (SHOX 5CA forward/reverse: CAT GTC ATA TAT ATA TGT GAT CC/GAC ACA GAA ATC CTT CAT AAA). PCR was performed using 2 U of Goldstar DNA Polymerase (Eurogentec, Cologne, Germany) in the buffer provided with the enzyme, 10 pmol/dNTP (MBI) and 75 mmol/MgCl2 in a Peltier Thermal Cycler (MJ-Research) (PTC-200) starting with denaturation (95°C (5 min)) followed by 35 cycles of amplification (95°C (30 s), 55°C (30 s) and 72°C (45 s)) and a final elongation step (72°C (5 min)). The PCR products were separated on polyacrylamide gels (10%) and analysed after silver staining.
FISH analysis
Biotinylated cosmid DNA was hybridised to metaphase chromosomes from stimulated lymphocytes of 247 participants under conditions previously described.26 FISH was performed using the SHOX-containing cosmid 34F5 (LLN0YCO3′M′34F5)5.
Mutation analysis
Mutation screening was performed by denaturing high performance liquid chromatography (DHPLC), using a WAVE DNA-Fragment Analysis System (Transgenomic Inc., Cheshire, UK). Mutation detection relies on differential binding to the column of homo- and heteroduplexes, which are generated by denaturing PCR products at 95°C for five minutes and cooling them slowly (–2°C per cycle, 44 cycles) to allow renaturing. Analysis was confined to the coding regions of the SHOX gene (exons 2–6). Primer sequences and PCR conditions were: exon 2 forward/reverse: CGC GGG GAG ACG CGC GCA TCC/GGC GCC GAA CCC CAG GAG GAG GGC; 63°C/65°C/66°C; exon 3 forward/reverse: GCC ACG TTG CGC AAA ACC TC/CCC GAG GAC CAG GCG ATG; 64°C/65°C/67°C; exons 4 and 5 were amplified together: exon 4 forward: GGG AGG CTG GGC TGG GTT C; 62°C/64°C/65°C; exon 5 reverse: GGA AGG GAG CAG CAG GTC C; 62°C/64°C/65°C; exon 6a forward/reverse: GTC CCC ATC CTG CGC CCT CAC CC/GCG CGG AGC CCG GGA GTC CG; 66°C/68°C/67°C).
Sequencing
Sequencing was performed on a MegaBACE sequencer (Amersham Bioscience, Piscataway, NJ, USA) using the DYEnamic ET terminator Cycle Sequencing Kit (CGE Healthcare, Freiburg, Germany) following the manufacturer’s protocol. Sequencing reactions were performed on both DNA strands. Sequences were analysed using the Clustal program (German Cancer Research Center, Biocomputing Facility HUSAR, Heidelberg, Germany).
Single nucleotide polymorphism (SNP)-based screening
Although the majority of DNA samples were analysed according to the methods described above, 275 participants had DNA analyses for SHOX deletions and mutations performed using a commercially available assay (SHOXDNAdx, Esoterix Inc, Calabasas Hills, CA, USA).27
Statistical analyses
Because the majority of participants originated from European countries, standard deviation scores (SDS) for height and the various anthropometric parameters were calculated using central European references.24 The exception was lower leg length, for which the reference range of Ross et al25 was used. Values were adjusted for the actual chronological age by linear interpolation. Body–mass index (BMI, kg/m2) was converted to SDS using the method of Cole.28 Comparisons between groups were performed using one-way analysis of variance (ANOVA) for continuous variables and Fisher exact tests for categorical variables at a two-sided significance level of 0.05.
For the development of a quantitative scoring system to assist in identifying patients for SHOX gene testing, logistic regression models were developed with confirmed SHOX deficiency (yes/no) as the dependent variable and various explanatory variables, including clinical diagnosis of LWS in the participant or relative(s), dysmorphic signs and anthropometric measures. The continuous variables were converted to categorical variables by applying defined cut-offs. For each group (SHOX and non-SHOX) the median for the particular variable was determined, and the mean of the two medians was then calculated and used as the cut-off. Variables were first tested one at a time, and included in the models only if there was a significant mean difference between the SHOX deficient and non-SHOX deficient groups. Next, the stepwise selection procedure was used to develop the models and the models were tested for lack of fit by applying the Hosmer–Lemeshow statistic. Scoring systems were developed, which included only those variables that contributed significantly to the logistic models as score items. The odds ratios generated by the logistic models were then rounded to the nearest integer and used as the score weights. Various score systems were applied to the study population and sensitivity, specificity, accuracy, and positive predictive values were calculated for different score point cut-offs.
RESULTS
A large cohort of 1608 children with short stature was screened for deletions and intragenic mutations of the SHOX gene. The physician-designated clinical diagnosis (LWS vs ISS) was collected at the entry of the study. Fifty-five participants (3.4%) were diagnosed with LWS; of the remaining 1553 participants 1534 were diagnosed as having ISS and for the other 19 participants there was no diagnosis or diagnoses other than LWS or ISS. SHOX defects were identified in 68 of the 1608 screened participants (4.2%), with 32/55 (58%) diagnosed with LWS and 34/1534 (2.2%) diagnosed with ISS, while two participants were not classified. The analysis for SHOX defects followed a step-wise process. To screen for a possible gene deletion, the first analysis determined the presence or absence of heterozygosity for a CA-repeat marker 14 kb (DXYS201) and CAII 34 kb upstream of the SHOX gene. When only a single allele size of these two markers was detected, implying homo- or hemizygosity, the next step was a FISH analysis using the SHOX-containing cosmid LLN0YCO3′M′34F5 to verify a putative deletion. Forty-eight participants (70.6%) had complete SHOX deletions and 4/68 (5.9%) had partial (small intragenic) deletions as indicated by a reduced FISH signal on one of the sex chromosomes. Intragenic mutations were detected in exons 2–6 of the coding sequence in 16/68 (23.5%) participants: 10 participants had missense mutations, 5 had nonsense mutations and 1 had a small duplication (table 1). Seven of the 16 mutations have not been reported previously. In vitro functional analyses of four missense mutations demonstrated reduced DNA binding (R168W), reduced dimerisation (L132V) or impaired nuclear translocation (A170P, R173H).29,30
Point mutations in the SHOX gene
Demographic and baseline clinical characteristics of the children with or without SHOX gene defects are shown in table 2. Although more males than females were enrolled in this study (952 (60%) male vs 656 (40%) female), the sex distribution of confirmed SHOX defects was reversed, with 54% of molecular defects being in females and 46% in males. As expected, children with SHOX deficiency had a significantly higher percentage of first-degree relatives with LWS or Langer syndrome than those without SHOX deficiency: fathers: 9.7% vs 0.5% (p<0.001), mothers: 26.6% vs 1.6% (p<0.001), siblings: 26.0% vs 2.0% (p<0.001). This finding is reflected by the decreased target height, which supports the concept of pseudoautosomal dominant inheritance of SHOX deficiency. SHOX testing was also performed in either one (n = 8) or both (n = 17) parents of 25 participants with SHOX deficiency. In 14 of 17 (82%) families in which both parents were tested, either the father or the mother had a SHOX defect; in no family did both parents have a SHOX defect. A SHOX mutation was found in 10 of 18 tested fathers (56%) and 8 of 24 tested mothers (33%). In all 25 cases in which DNA was also available for family members the mutations co-segregated with the phenotype of LWS or short stature.
Demographic and baseline data of patients with (SHOX) and without (non-SHOX) a deletion or mutation in the SHOX gene and patients with Turner syndrome
Investigators were instructed to examine the participants at screening for the presence of a specified list of dysmorphic signs (table 3). A significant number of participants with a pre-study diagnosis of ISS who were subsequently confirmed to have a SHOX defect had some dysmorphic signs typical of LWS. Only a few signs such as micrognathia, high-arched palate, short forearm and Madelung deformity were significantly more frequent in participants with a baseline clinical diagnosis of LWS (table 3). For children with a clinical diagnosis of LWS there was no statistically significant difference in the frequency of dysmorphic signs between children who were subsequently confirmed to have a SHOX defect (n = 32) and those without an intragenic mutation of the SHOX gene (n = 23; data not shown). For participants with a clinical diagnosis of non-syndromic short stature (“ISS”) a number of clinical signs were more prevalent in those with a molecularly confirmed SHOX defect (n = 34) than in those without a SHOX defect (n = 1500; table 4). Therefore, these physical signs (shortening or bowing of the forearm, dislocation of the ulna at the elbow, bowing of the tibia and appearance of muscular hypertrophy) appear to represent clinical indicators for SHOX deficiency in short children. In addition, participants with proven SHOX defects had significantly greater mean body–mass index (BMI) standard deviation score (SDS) than those without.
Dysmorphic signs in patients with confirmed SHOX deficiency
Dysmorphic signs in patients with non-syndromic short stature (ISS) excluding patients with Léri-Weill syndrome, with unknown diagnosis or with a diagnosis other than ISS
Because SHOX haploinsufficiency is also believed to underlie the short stature and skeletal anomalies of Turner syndrome, frequencies of dysmorphic signs were compared between participants with Turner syndrome (n = 33) and those with (n = 68) or without (n = 1540) confirmed SHOX deficiency (table 5). While the majority of dysmorphic signs were not different in frequency between the SHOX-deficient and Turner syndrome groups, there were a few notable exceptions. High-arched palate and cubitus valgus were substantially more common in Turner syndrome, whereas short forearm or short lower leg, Madelung deformity and muscular hypertrophy were markedly more prevalent in the SHOX-deficient group.
Dysmorphic signs in patients without SHOX deficiency (non-SHOX), with SHOX deficiency (SHOX) and with Turner syndrome; both the non-SHOX and SHOX groups include patients with Léri-Weill syndrome
To obtain an objective assessment of body proportions a number of anthropometric measurements were performed. Values were adjusted for standing height by calculating the ratio (%) of the anthropometric measure divided by height (table 6). Participants with SHOX deficiency had a significantly greater sitting height and forearm, upper arm and upper leg circumference adjusted for standing height. In contrast, height-adjusted arm span, forearm length and lower leg length were significantly shorter in participants with SHOX deficiency compared with those without. These findings underline the differences in frequency of dysmorphic signs between the groups with and without SHOX deficiency and suggest mesomelic disproportion and appearance of muscular hypertrophy as the most indicative features of SHOX deficiency. There were no significant differences in dysmorphic signs or anthropometric measures between participants with a missense mutation within (n = 6) or outside (n = 5) the homeobox (data not shown).
Anthropometric measures in patients with (SHOX) and without (non-SHOX) SHOX deficiency
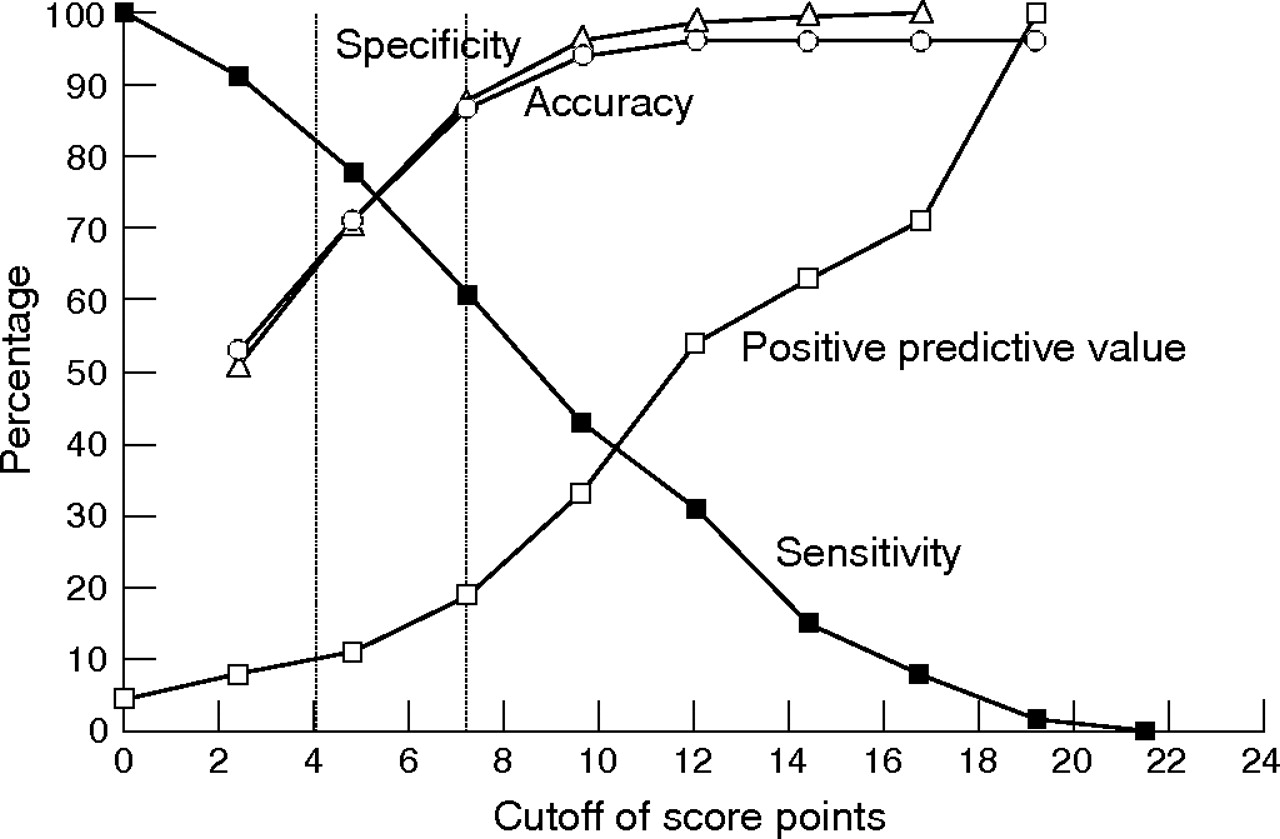
Although the above analyses elucidate the spectrum of phenotypic findings in children with various forms of SHOX deficiency, the variability of the phenotype makes it difficult to decide whether or not SHOX testing is warranted for an individual child. Therefore, to provide evidence-based scoring systems for identifying the most appropriate children for SHOX gene testing on clinical grounds, various logistic regression models were developed with SHOX deficiency (yes/no) as the dependent variable. Combinations of variables, including diagnosis (LWS in subject or relatives), dysmorphic signs and anthropometric variables that were significantly different between participants with or without SHOX deficiency, were included in the models as explanatory variables, and odds ratios were calculated. The diagnosis of LWS or the presence of Madelung deformity in the subject or in a family member resulted in very high odds ratios in the models. Therefore, to focus on the clinically more challenging subjects with non-syndromic short stature (“ISS”), these strong indicators of SHOX deficiency were excluded from the models. The scoring system presented in table 7 is based on the final logistic regression model, using rounded odds ratios as weighted score points. There was no evidence of lack of fit of the underlying logistic model (lack of fit P = 0.81). The suggested scoring system includes three anthropometric variables (arm span/height, sitting height/height and body mass index (BMI)) and five dysmorphic signs. This scoring system, in which a maximum of 24 score points can be attained, was applied to the study population to evaluate the performance of the score. The dependence on various score point cutoffs of sensitivity, specificity, accuracy and positive predictive value is shown in Figure 1. At a cut-off of >4 points, sensitivity was 71% and the positive predictive value was 11%; at >7 points the sensitivity dropped to 61% while the positive predictive value increased to 19%.
Scoring system for identifying patients that qualify for short-stature homeobox containing gene (SHOX) testing based on clinical criteria

{kind=link}
Dependence of sensitivity, specificity, accuracy and positive predictive value on cut-off score point; the vertical lines represent cut-offs of >4 and >7 score points, respectively.
DISCUSSION
This multi-centre, multi-national study was performed to define a comprehensive clinical and molecular spectrum for SHOX haploinsufficiency. Whereas the involvement of the SHOX gene in short stature is well established, there is little prospective quantitative data addressing the more subtle clinical features and genotype-phenotype relationships in participants with SHOX deficiency. We therefore evaluated the growth and skeletal features of a large cohort of children with short stature and performed a detailed analysis of their anthropometric measures and dysmorphic features, comparing the findings in participants with proven SHOX defects to those in participants without demonstrable molecular SHOX defects. We also compared the clinical features in children with isolated SHOX deficiency to those seen in another model of SHOX haploinsufficiency children with Turner syndrome; within the cohort with isolated SHOX deficiency (ie, excluding participants with Turner syndrome), we compared the findings of participants with the classical LWS phenotype with those participants with more subtle phenotype, diagnosed before entry to the study as ISS. Finally, we defined diagnostic criteria that will allow clinicians to identify patients most likely to have a SHOX gene abnormality and to target SHOX testing to these patients.
SHOX mutations were identified in 4.2% of 1608 children studied. However, this cohort was enriched for participants with LWS, in whom the frequency of SHOX defects is particularly high; when these participants were excluded and only the participants with non-syndromic (or idiopathic) short stature were included, the frequency of SHOX deficiency was 2.2%. This frequency is similar to the rates reported in previous smaller studies in independent samples of children with ISS using similar genetic and molecular methodologies7,10; however, it is lower than frequencies reported in other studies.8,9,11 These variable rates of detection of SHOX defects may in part relate to methodological differences or to the different population studied.11,31,32 We also did not screen for putative enhancer mutations (“downstream deletions”) nor single exon deletions. It is therefore likely that intragenic mutations due to splice defects in the introns, regulatory mutations or single exon deletions have been missed.
Short stature is a more frequent reason for referral to a pediatric endocrine clinic for boys than for girls. Therefore, it was not surprising that 60% of our study participants were boys. However, it is interesting to note that SHOX gene defects were confirmed in a greater proportion of tested girls than boys (54% vs 46%). This finding is consistent with the greater reported prevalence of LWS in females,33 hypothesised to result from an effect of estrogen at the SHOX-deficient growth plate.20 If true, this effect may have led to an ascertainment bias toward females with clinically significant skeletal anomalies.
Consistent with previous reports, the majority (52/68; 77%) of mutations detected were complete or partial gene deletions.7,10,11,21 The relatively high frequency of deletions has been attributed to the structural composition of the pseudoautosomal region, which may promote recombination, thereby predisposing to deletion events.34,35 Notably, the majority of deletions have been transmitted through the paternal allele suggesting that the high recombination rate during male meiosis is accountable for this phenomenon.36
Although the majority of the SHOX gene defects in this study were large scale deletions, there were nevertheless a number of novel intragenic mutations that may elucidate some aspects of SHOX structure or function. The spectrum of intragenic mutations in the SHOX gene in this and other studies is highly heterogeneous. Including the seven novel mutations reported here, more than 60 different mutations have been identified, distributed throughout the coding regions of the gene (http://www.shox.uni-hd.de). The majority of intragenic mutations are missense mutations, and all missense mutations detected in this study affect amino acids conserved between human, chicken and fish, highlighting the functional importance of these affected residues. None of these missense mutations was detected in unaffected parents tested in this study or in 100 unaffected control individuals screened outside this study. While the fact that the missense mutations segregate with the phenotype provides evidence of the functional role of the mutations, definitive proof requires in vitro demonstration of disturbed activity of the encoded transcription factor. Functional consequences have been demonstrated for the mutants R168W, L132V, A170P and R173H with respect to DNA binding, dimerisation or nuclear translocation of the SHOX protein.29,30 However, the effect of Q112P, K116E, insH229-L232 and X226R on SHOX activity is unknown.
Although it might have been expected that children with molecular SHOX gene defects would have had more severe short stature than those with normal SHOX genes, we found no difference in the degree of short stature between those with or without an identifiable SHOX gene defect; mean height was –2.6 SDS in both groups. In fact, the most striking anthropometric difference between SHOX-deficient and non-SHOX deficient participants was the 1.0 SDS greater body mass index (BMI) in the SHOX-deficient group (p<0.001). This finding is interesting in light of the fact that participants with a working diagnosis of ISS (who comprised half of the SHOX-deficient group in our study) typically have BMI values in the low to low-normal range, perhaps due to poor eating behaviour.37 However, rather than reflecting obesity, the above average BMI for age and sex in SHOX-deficient children likely reflects segmental disproportion with rather normal trunk size but short legs. Moreover, the relative increase in limb circumferences (discussed below) suggests increased muscle mass or muscular hypertrophy, which may translate to a greater body mass compared with the significantly reduced height of affected individuals, leading in turn to an increase in BMI.
Apart from the increased BMI, the most reliable clinical indicators of SHOX deficiency were disproportionate growth, with shortening of the limbs compared with the trunk and bowing of the forearm or lower leg. The limb shortening was evidenced by reductions in the ratios of arm span and forearm length to height and an increase in sitting height/height ratio in the SHOX-deficient vs SHOX-sufficient group; furthermore, arm span and forearm length vs height were all reduced in the SHOX-deficient group compared with reference values for age. These findings are consistent with other reports.10,38 All limb circumferences (upper arm, forearm, upper leg and lower leg) were relatively increased compared with height in SHOX deficient vs non-SHOX deficient participants, and were increased relative to reference values for all measures except upper leg circumference/height. These findings of increased limb circumferences were manifest clinically as the appearance of muscular hypertrophy, which was significantly more common in the SHOX-deficient participants, as previously reported.21 Although it is possible that the apparent muscular hypertrophy may result from unfavourable lever proportions of the limbs, it may also represent a genuine developmental phenomenon due to SHOX haploinsufficiency. Notably, these findings were present in a significant number of participants with the “ISS” phenotype as well as those with LWS, indicating that these diagnoses represent a continuum, rather than discrete entities.
The clinical features of SHOX deficiency beyond the effects on long bone length, appear to represent changes in the curvature or angularity of certain bones. As examples, the following features were present in a significantly greater proportion of participants with SHOX deficiency than those without: forearm and tibial bowing, cubitus valgus and genu valgum, Madelung deformity and ulnar dislocation at the elbow. In addition, it should be mentioned that lower leg and forearm x rays can be very helpful to detect deformities, which are clinically not obvious. Furthermore, as reported previously, a number of these features were also seen with increased frequency in the Turner syndrome group, including high-arched palate, forearm bowing, cubitus valgus, ulnar dislocation, and a variety of other dysmorphic signs. Although the phenotypic differences between SHOX-deficient participants with LWS vs “ISS” were subtle, there were a number of features that were significantly more common in children with LWS than in those with the baseline clinical diagnosis of ISS, including high-arched palate, Madelung deformity, micrognathia and shortening and/or bowing of the forearm. These alterations in the patterns of bone growth likely reflect the disorganisation of the growth plate that has been reported in association with SHOX deficiency.14 Despite some differences between the diagnostic groups within this study in terms of frequency of the various clinical findings, it has become clear that there is no single phenotype of SHOX deficiency, but rather that molecular defects of the SHOX gene may manifest a widely varying spectrum of clinical severity, most likely reflecting other generic influences on the expression of the genetic disorder.
The presence of LWS or Madelung deformity in an individual participant or in a first degree relative in this study was an overwhelming indicator for the presence of SHOX deficiency. Therefore, all children with these clinical diagnoses should undergo formal SHOX gene molecular analysis. However, the same is not true for the milder forms of SHOX deficiency detected in this study. In participants with non-syndromic forms of short stature the key clinical features of SHOX deficiency, evidenced by significant differences between the SHOX-deficient and non-SHOX-deficient groups, were increased BMI, short limbs relative to overall height (eg, short forearm: 21% vs 7%; p = 0.006), evidence of muscular hypertrophy (27% vs 9%; p = 0.003), and increased bowing or angularity of various bones, particularly those of the lower leg (tibial bowing: 15% vs 3%; p = 0.002) and forearm (12% vs 3%; p = 0.027).
Although this study undertook a thorough assessment of the phenotype of children with SHOX deficiency, we were unable to determine any clear correlation between the specific SHOX gene defect and the clinical features. For example, no consistent differences could be discerned between participants with complete SHOX gene deletions and those with missense mutations resulting in conservative amino acid substitutions. Furthermore, although SHOX haploinsufficiency is present in both LWS and Turner syndrome and might be expected to result in similar skeletal features, the findings of high-arched palate and cubitus valgus were significantly more frequent in children with Turner syndrome, whereas children with isolated SHOX deficiency had a significantly greater frequency of Madelung deformity, short forearm, short lower leg and appearance of muscular hypertrophy. The basis for these phenotypic differences is speculative: one suggested explanation is that estrogen action at the growth plate in girls with isolated SHOX deficiency (ie, normal gonadal function) may exacerbate the effects of SHOX haploinsufficiency20; alternatively, it may be hypothesised that haploinsufficiency of additional X-chromosomal genes in girls with Turner syndrome may mitigate the effects of SHOX deficiency to some degree. In addition to the absence of a genotype-phenotype relationship for SHOX gene defects, there also appear to be other defects that manifest an indistinguishable clinical phenotype, evidenced by the fact that a significant minority of participants with typical dysmorphic signs of LWS had no detectable SHOX gene defect in this and other studies.8,9,11 The absence of a clear molecular basis for the LWS phenotype in a significant proportion of participants suggests that a similar phenotype can be caused either by intragenic mutations that may escape detection due to their location in regulatory regions of the gene, or to defects at a different, but perhaps related, genetic locus.
To provide a quantitative approach to determining the most appropriate candidates for SHOX genetic testing, we integrated the clinical and anthropometric findings reported here into a pragmatic scoring system that optimises the selection of patients who are less clinically obvious than those with LWS and Madelung deformity. The score combines three anthropometric variables (arm span/height ratio, sitting height/height ratio and BMI), with five clinical variables (cubitus valgus, short forearm, bowing of forearm, muscular hypertrophy and dislocation of the ulna at the elbow), each of which represents at least two points in the score system (table 7). The system was then examined for sensitivity, specificity, positive predictive value and accuracy using various total score values. On the basis of these analyses, testing for SHOX deficiency is recommended for participants with a score greater than four or seven out of a total possible score of 24. The decision on whether to choose the increased threshold score needs to take into account the decrease in sensitivity, which means more SHOX-deficient patients will be missed, versus the increase in positive predictive value, which means more patients will be tested positive for a SHOX defect.
In summary, SHOX defects represent a relatively frequent cause of short stature, particularly in children with evidence—which may sometimes be quite subtle—of shortening and/or bowing of the forearms and lower legs. The presence of some of the typical physical findings of LWS in children with the initial clinical diagnosis of ISS indicates that a very careful physical examination, including measurement of body proportions, must be performed before making the diagnosis of ISS. In addition, x ray analysis of the lower leg and forearm can be very important when distinguishing between ISS and LWS. The presence of any combination of the following physical findings: reduced arm span/height ratio, increased sitting height/height ratio, above average BMI, Madelung deformity, cubitus valgus, short or bowed forearm, dislocation of the ulna at the elbow, or the appearance of muscular hypertrophy, should prompt the clinician to obtain a molecular analysis of the SHOX gene.
Acknowledgments
We thank the patients and their relatives for their participation in this study. We also thank the following colleagues for their enthusiastic support of this research: JS Fuqua (Indianapolis, IN, USA), NT Mauras (Jacksonville, FL, USA), R McVie (Shreveport, LA, USA), T Moshang (Philadelphia, PA, USA), P Saenger (New York, N.Y., USA), C Deal (Montreal, Canada), N Gagne (Montreal, Canada), M Lawson (Ottawa, Ontario, Canada), L Stewart (Vancouver, BC, Canada), D Wherrett (Toronto, Canada), M Bettendorf (Heidelberg, Germany), A Grüters-Kieslich (Berlin, Germany), P H Heidemann (Augsburg, Germany), E Keller (Leipzig, Germany), K Mohnike (Magdeburg, Germany), M B Ranke (Tübingen, Germany), E Schönau (Cologne, Germany), W Schönberger (Mainz, Germany), G Simic-Schleicher and S Spranger (Bremen, Germany), W G Sippell (Kiel, Germany), J-C Carel (Paris, France), P Chatelain (Lyon, France), M Colle (Bordeaux, France), Y Lebouc (Paris, France), J Leger (Paris, France), B Leheup (Vandoeuvre Les Nancy, France), P Limal (Angers, France), P Sultan (Montpellier, France), M Tauber (Toulouse, France), J Argente (Madrid, Spain), M de los Desamparados and R Sanchez (Madrid, Spain), J del Valle (Sevilla, Spain), P Martul (Vizcaya, Spain), L L S Drop (Rotterdam, The Netherlands), H M Reeser (The Hague, The Netherlands), J P Bourguignon (Liège, Belgium), M Craen (Gent, Belgium), J De Schepper (Brussels, Belgium), I François and F DeZegher (Leuven, Belgium), C Heinrichs (Brussels, Belgium), M Maes (Brussels, Belgium) R Rooman (Edegem, Belgium), J Lebl (Prague, Czech Republic), J Zapletalova (Olomouc, Czech Republic), M Dumic (Zagreb, Croatia), F Peter (Budapest, Hungary), G Soltesz (Pecs, Hungary), A Lewinski and Hilczer (Lodz, Poland), T Romer (Warszawa, Poland), V Peterkova and A Vitebskaya (Moscow, Russia), A Buyukgebiz (Izmir, Turkey), H Gunoz (Istanbul, Turkey), P Isguven (Istanbul, Turkey), G Ocal (Ankara, Turkey), V and A Khaldikar (Pune, India), N Shah (Mumbai, India), U Sriram (Mylapore, India).
REFERENCES
Footnotes
-
Published Online First 20 December 2006
-
Competing interests: None declared.