Article Text

Abstract
The Xq28 duplication involving the MECP2 gene (MECP2 duplication) has been mainly described in male patients with severe developmental delay (DD) associated with spasticity, stereotypic movements and recurrent infections. Nevertheless, only a few series have been published. We aimed to better describe the phenotype of this condition, with a focus on morphological and neurological features. Through a national collaborative study, we report a large French series of 59 affected males with interstitial MECP2 duplication. Most of the patients (93%) shared similar facial features, which evolved with age (midface hypoplasia, narrow and prominent nasal bridge, thick lower lip, large prominent ears), thick hair, livedo of the limbs, tapered fingers, small feet and vasomotor troubles. Early hypotonia and global DD were constant, with 21% of patients unable to walk. In patients able to stand, lower limbs weakness and spasticity led to a singular standing habitus: flexion of the knees, broad-based stance with pseudo-ataxic gait. Scoliosis was frequent (53%), such as divergent strabismus (76%) and hypermetropia (54%), stereotypic movements (89%), without obvious social withdrawal and decreased pain sensitivity (78%). Most of the patients did not develop expressive language, 35% saying few words. Epilepsy was frequent (59%), with a mean onset around 7.4 years of age, and often (62%) drug-resistant. Other medical issues were frequent: constipation (78%), and recurrent infections (89%), mainly lung. We delineate the clinical phenotype of MECP2 duplication syndrome in a large series of 59 males. Pulmonary hypertension appeared as a cause of early death in these patients, advocating its screening early in life.
- X-linked
- genetic counselling
- MECP2duplication syndrome
- MECP2gene
- Xq28 duplication
- facial dysmorphism
Statistics from Altmetric.com
Introduction
The MECP2 gene encodes an essential epigenetic regulator in postnatal brain development involved in transcriptional activation as well as repression of many genes.1–3 Loss of function mutations of MECP2 in females result in classic Rett syndrome, a progressive childhood neurodevelopmental disorder, while hemizygous mutations found in males are often lethal.4 Different MECP2 pathogenic variations have also been described in male patients with a phenotype ranging from severe neonatal encephalopathy to non-specific moderate intellectual disability (ID).5 6 An increased dosage of methyl-CpG binding protein-2 (MeCP2) resulting from a duplication of the Xq28 region including MECP2 (MECP2 duplication) (MIM 300260), leads to a severe neurodevelopmental disorder in males.7–9 This disorder generally includes early hypotonia, progressive lower limbs spasticity, very poor to absent speech, severe epilepsy, stereotypic movements and recurrent infections. Since the initial report, around 160 male patients have been reported. This has led to the description of a clinically recognisable syndromic ID.10–12 It has been shown that a small proportion of carrier females could be symptomatic, with a non-specific and mild developmental delay (mostly) or a severe phenotype.13–17 In a previous study, we reported a series of 20 symptomatic females carrying a de novo interstitial MECP2 duplication (6 novel and 14 reported patients). Thirty-seven per cent had inherited their duplication from their mother, including three mildly, one moderately and three severely affected patients. No correlation was shown between X chromosome inactivation (XCI) in the blood or duplication size and the severity of the phenotype. These findings confirmed that an abnormal phenotype, even severe, can be a rare event in females born to asymptomatic carrier mothers, making genetic counselling difficult in couples at risk.17
Previous studies in mice also showed that MECP2 dosage plays a major role in postnatal neurodevelopment. Mice with twice the endogenous level of MeCP2 appear normal until 10–12 weeks of age, after which they display forepaw stereotypies, impaired coordination, seizures, hypoactivity and spasticity.18 It has also been shown that increased MeCP2 dosage in mice leads to increased synaptic numbers, having therefore a major impact on the formation of excitatory synapses.19 In the same way, it has been elegantly demonstrated that cortical neurons derived from different induced pluripotent stem cells (iPSC) lines of patients with MECP2 duplication syndrome have increased synaptogenesis and dendritic complexity and that their neuronal network synchronisation was altered.20 These findings contribute to the understanding of the underlying pathways that lead to the very severe neurodevelopmental phenotype of this disease. Furthermore, a more recent study showed that antisense oligonucleotides treatment could rescue many features in adult symptomatic transgenic MECP2 duplication mice, which appears to be a very promising therapeutic approach.21
Although the size of the duplications differs from one patient to another, and despite the involvement of several other brain-expressed genes than MECP2, such as SLC6A8 (MIM 300036), L1CAM (MIM 308840), FLNA (MIM 300017) and GDI1 (MIM 300104)—which are responsible for various neurodevelopmental disorders—it has been demonstrated that the increased dosage of MECP2 is sufficient to explain the core phenotype of MECP2 duplication syndrome.10 Indeed, several affected patients harbouring the smallest duplications, only including IRAK1 and MECP2, presented with developmental delay, epilepsy, spasticity and recurrent lung infections.8 22 23
The exact prevalence of MECP2 duplication syndrome remains unknown, although different screening attempts have estimated that it may explain about 1% of severe X-linked intellectual disability (XLID) cases.24 Very few large series have been reported to date, in particular those regarding the morphological phenotype. Following active data collection from 24 different French clinical genetic departments and 17 cytogenetic and molecular laboratories, we now report a large French series of affected males with an intrachromosomal Xq28 duplication involving MECP2. We describe the clinical phenotype with a focus on the morphological and neurological features of this disorder. We also review the patients reported to date in the literature and report more clinical features.
Patients and methods
Data collecting
Patients carrying an Xq28 duplication involving MECP2 (MECP2 duplication) were recruited through a French collaborative study conducted from 2012 to 2016, for phenotypical delineation and genotype-phenotype correlation purposes (XLID Research Project 2008–2016, French Ministry of Health). For each patient included, medical data were taken both retrospectively from medical records and as far as possible, cross-sectionally during a dedicated medical consultation. During the latter, the physician asked the parents about the medical history (including the motor skills, behaviour, way of life, kinds of re-education, etc), performed a clinical examination, filled out a detailed questionnaire and took morphological pictures. Since most of the patients have a poor intellectual functioning, no standardised psychometric test could be performed. The degree of ID was determined on the basis of adaptive skills (ambulation, motor development, communication, sociability, daily life autonomy), in accordance with the Diagnostic and Statistical Manuel of mental Disorders, fifth edition.25 Additional investigations were only performed if beneficial for the patient, accounting for missing clinical and radiological data.
Cytogenetic and molecular analyses
The duplications were detected using a pangenomic (Array Comparative Genomic Hybridization (Array-CGH)) or a targeted approach (Multiplex Ligation-dependent Probe Amplification (MLPA) or quantitative PCR (qPCR) analyses), which were performed in 16 French cytogenetic and molecular laboratories and one Belgian laboratory (Leuven). They were performed on genomic DNA isolated from peripheral blood samples. Regarding Array-CGH, the platform used for most of the patients was the Human Genome CGH Microarray 150K or 180K from Agilent according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, California, USA). MLPA was performed using the method described by Schouten et al in case the clinicians suspected the MECP2 duplication syndrome.26 The duplications were confirmed by fluorescence in situ hybridization (FISH) with various specific probes on chromosome preparations from leucocyte cultures or by qPCR using standard protocols. Parental analyses were performed using FISH or qPCR. Genomic positions were relative to human genome GRCH37/hg19.
All patients’ families provided written informed consent and all procedures performed in the studies were done in accordance with the ethical standards of the institutional research committee and with the Declaration of Helsinki.
Results
Study population
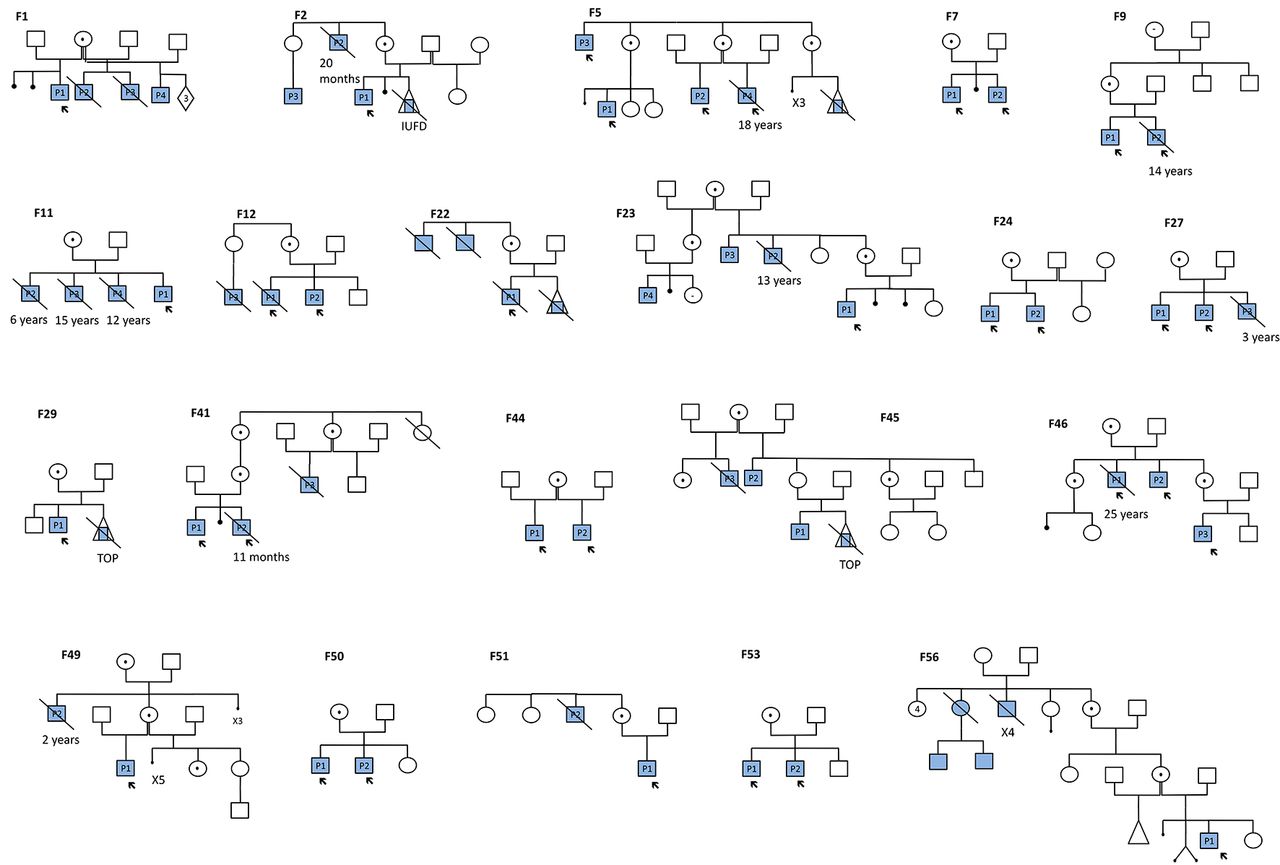
One hundred and one cases, including 91 males and 10 females (including twins), were identified with a MECP2 duplication in the context of their diagnostic workup for ID. Among the 91 males, the duplications resulted from interstitial (intraC) Xq28 duplication in 86 cases (54 families) and from a complex chromosomal aberration, such as X-autosome translocation or insertion of the Xq28 region into an autosome, in five cases. Twenty-three out of the 86 patients (27%) died before the age of 25 years (11 months to 24 years, mean age: 9.5 years). Among the 11 patients for whom the cause of the death was known, 8 (73%) died from respiratory infections, 2 (18%) from a pulmonary hypertension and 1 (9%) from status epilepticus. Seventy-three out of the 83 for whom we had the information (88%), had inherited their duplication from their mothers. The pedigrees of the 19 families with an X linked inheritance are illustrated in figure 1. The present study reports only on the 86 males with intraC MECP2 duplication, with a focus on the 59 patients for whom we were able to collect clinical and morphological data, including photographs. Twenty-five were examined by the same physician (SEC), which could help highlight recurrent clinical features that were often overlooked.

Family trees. The full squares represent the affected males
Cytogenetic and molecular data
Duplications were detected using genomic microarrays in 25 out of the 54 families (46%) and targeted analysis in 29/54 families (54%). The analysis of the 46 families in which the duplication breakpoints were available showed that the Xq28 duplications were different in size and breakpoint between patients as illustrated in online supplementary figure 1. We observed a recurrent distal breakpoint in nine patients (153609163 bp) suggesting a susceptibility region in Xq28 to such a rearrangement, and 85% (39/46) of the duplications shared a common region of 1.5 Mb located between position 152 485 412 and position 153 949 811 (hg19) (online supplementary figure 1 and table 1). The mean size of the duplications was 1856 kb (range: 102–16 704 kb), and the main genes included in the duplications were MECP2, IRAK1, GDI1, FLNA and L1CAM. Since the patients with the smallest duplications did not have a milder neurological phenotype, we could not show an obvious correlation between the size of the duplications and the severity of the phenotype. The FMR1 gene was involved only in the three patients harbouring the largest duplications (online supplementary figure 1 and table 1). XCI studies showed that all of the carrier mothers for whom data were available had completely skewed XCI (XCI ratio >80:20) (data not shown).
Supplemental material
Clinical data
We were able to collect clinical data for 59 out of the 86 male patients harbouring an intraC MECP2 duplication. Among them, nine (15%) died before the age of 25 years. The mean age at examination was 11.7 years (range: 2 months to 48 years), with 13 adult patients (age ≥18 years). The clinical data are summarised in tables 1 and 2 and online supplementary table 2.
Supplemental material
Summary of the main facial and limb morphological features from the present cohort
Comparison of the main clinical features from the 59 patients of the series
Perinatal period
The mean term of birth was 39 weeks of gestation (WG) (range: 32–41). Four patients were born prematurely (<37 WG). Birth parameters were within the normal range with a mean weight at −0.1 SD score (SDS) (−2.6 SDS; +3.5 SDS), mean height at −0.7 SDS (−4.5 SDS; +1.9 SDS) and mean head circumference at +0.1 SDS (−2.5 SDS; +4.5 SDS). The Apgar score was abnormal (<8) for 10% (5/50) of the newborns and 20% (10/50) were admitted to either a neonatology department or an intensive care unit. Later, 55% (32/58) of the patients had complications during the neonatal period, including early hypotonia (11/58), feeding difficulties (15/58) or infection (4/58) (online supplementary table 2).
Growth parameters and craniofacial features
The growth parameters were mainly in the normal range. Mean occipitofrontal circumference (OFC) was +0.2 SDS (−3.4 SDS; +4.8 SDS). Four out of 53 (8%) patients had microcephaly (OFC <−2 SDS) and 9/53 (17%) were macrocephalic (OFC >+2 SDS) (table 1). The mean height was −0.4 SDS (−5.2 SDS; +2.8 SDS) and the mean weight was +0.4 SDS (−4.7 SDS; +5.9 SDS) (table 2). Most of the patients had particular facial features (51/55), summarised in table 1 and illustrated in figures 2 and 3. We noted that most of the affected males had a similar facial gestalt, characterised by a midface hypoplasia (93%), deep set eyes (58%), hypertelorism (45%), a small (70%) open mouth (98%) with a thick lower lip (78%) and intermittent drooling (80%), large prominent ears (85%), narrow (64%) and prominent (76%) nasal bridge with anteverted nostrils (52%). Hair was often thick and dense (91%) with a sparse area on the lateral part of the forehead (69%) (figure 2, patients F53P2, F5P1, F55P1), which led to a temporal spike aspect, and the eyebrows were often sparse (54%) (figure 2, patients F29P1, F40P1, F5P4) with a synophrys sometimes apparent (26%). A bilateral epicanthus was noted in 41% of the patients. We noted that patients with a light skin phototype had particularly pale and bright blue eyes. Other recurrent features were dental shape and/or eruption abnormalities in 94% of the series, including prominent central incisors (50%), and persistence of the deciduous teeth (88%), often requiring multiple tooth extractions (table 1, figures 2 and 3).

Facial phenotype of the patients with an interstitial Xq28 duplication involving MECP2. Patients share common facial features including an open mouth with a thick lower lip and intermittent drooling, large ears, midface hypoplasia, deep set eyes with frequent divergent strabismus, sparse eyebrows, narrow and prominent nasal bridge, dense and thick hair with an anterior sparse area.

Focus on the lower part of the face in patients with an interstitial Xq28 duplication involving MECP2. Note the small mouth and the open mouth appearance with a convex upper lip during early infancy (A, B), and the prominent central incisors after childhood (C).
We found that 62% of the patients had abnormally small feet which were often flat with a valgus deformation (74%). The hands showed tapered fingers in 36/39 (92%) patients and a camptodactyly of several fingers was noticed in few cases (figure 4B). Frequent vasomotor problems were noted (48%) leading to moist and red hands and/or feet (figure 4C). We also noted an apparent venous network (livedo) of the limbs in 34/39 (87%) of the children, which tended to be less evident after adolescence (figure 4D) (table 1).

Common habitus with a flexion of the trunk and the knees (A), slender hands with tapering fingers and camptodactyly in few cases (F9P1, F44P1) (B), vasomotor troubles in the hands which appear red and wet (C), apparent venous network (livedo) of the limbs (D).
Interestingly, in the 45 patients for whom photographs were available from early infancy (<3 years) to school age (6–14 years) or adulthood, we were able to observe, as often in syndromology, an evolution of the facial features with ageing. In early infancy, the face looked mostly round with a small chin and full cheeks and the mouth already appeared small, with downturned corners, a convex upper lip and open appearance with frequent drooling visible (figures 3 and 5). With time, the face became elongated, with a prominent chin and a larger mouth with sometimes prominent central incisors. The nose was initially particularly small with anteverted nostrils and changed with frequent prominent nasal bridge and tip. Facial features tended to become coarser and eyes appeared deeper after adolescence and during adulthood, although facial dysmorphism was very mild in some patients (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Evolution of the phenotype with time. Evolution of the facial features from early infancy to school age or adulthood (from left to right). Note the changing facial phenotype from round face with small mouth and nose to elongated coarser face with prominent nose and ears and thick lower lip.
Hearing and vision
Seven out of 23 (30%) patients who were assessed had mild-to-moderate hearing loss (table 2). The type of the deafness was specified in three cases only (two patients had conductive deafness in a context of numerous otitis and one had perceptive deafness). A divergent strabismus was frequent (41/54, 76%) and could be related to visual impairment since 54% (21/39) of the patients had hypermetropia. Three patients had corneal anomalies (scars and sclerocornea). A ptosis was noted in 30% of the patients.
Neurodevelopmental and orthopaedic features
During childhood, almost all patients had early hypotonia (57/58) (online supplementary figure 2) and all had global DD (59/59). Motor skills were severely affected as 12/56 (21 %) were unable to achieve ambulation without support. For the remaining patients, the mean age of independent walking was delayed (3.6 years (range: 1.8; 5.4)). Among them, seven lost the ability to walk. Patients developed orthopaedic symptoms that could be, at least partially, the consequences of lower limbs spasticity and hypotonia. One of the main features was kyphoscoliosis, present in 53% of them (23/43). Another striking feature was a lower limbs spasticity leading to a singular habitus with a flexion and sometimes contractures of the ankles, the knees and the trunk. This spasticity was not often associated with pyramidal signs as Babinski sign or hyper-reflexia but led to an abnormal pseudoataxic gait with a degree of instability and a broad-based stance (figure 4A, table 1). We hypothesise that this unstable gait is partly due to an abnormal development of the proprioceptive pathways. This leads to a shift of the centre of gravity of the body which is partially compensated by the flexion attitude.
Supplemental material
![[SP4.jpg]](https://jmg.bmj.com/content/jmedgenet/55/6/359/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
Epilepsy
Seizures were seen in 59% (35/59) of individuals with a mean onset around 7.4 years of age (0.4–35). Seizure types were generalised tonic-clonic seizures (19/35), absences (5/35), partial complex (6/35), atonic (12/35) and myoclonic seizures (8/35). Sixty-two per cent (21/34) had drug-resistant epilepsy. In many patients, active epilepsy did not stop in late adolescence, since 12 of the 14 oldest living patients of our cohort (16–49 years), still experience seizures, including 10 who were drug-resistant. Among 31 patients for whom data were available, a neurological regression was observed after seizure onset in 12 patients (39%), including 11 patients with drug-resistant epilepsy. Among the 42/54 (78%) patients with the most severe developmental delay (5 youngest patients excluded), 14 (33%) did not develop epilepsy.
Communication and behaviour
All of the patients had ID, which was severe in most of them. A majority of patients were dependent for daily life needs (dressing, bathing, toileting, etc). Only five children became toilet trained but two of them lost this skill. Among the patients for whom data were available, the majority did not develop speech (32/57, 56%) or had just babbling (4/57, 7%) or few words (20/57, 35%). However, we observed that for a few patients who had highly active familial and paramedical stimulations and re-education, a communicative strategy could be elaborated, including the use of pictograms. Many patients had stereotypies (49/55, 89%), including flapping, midline hands movements, head movements and 33/46 (72%) had bruxism. These stereotypic movements tended to appear during school age and persisted, in a variable way, throughout life. Regarding the autistic features, we could observe that, even though most of the patients had stereotypies and poor to absent language, with regression of skills for some, a considerable proportion demonstrated several interactions during the consultation, without social withdrawal, they smiled frequently and were interested in trying to carry out simple tasks.
In addition, a majority of parents reported to us decreased pain sensitivity (29/37, 78%) (table 2). The parents of two children who had broken their leg mentioned that they had not shown any pain.
Other medical issues
Not surprisingly, one of the most frequent features was chronic constipation, which was found in 78% of the patients (43/55), including in some of them, subobstruction episodes or even chronic intestinal pseudo-obstruction (CIPO) syndrome. When rectal biopsies were performed they were normal. Most of the patients had early feeding difficulties (34/56, 61%). They were partially due to an important gastro-oesophageal reflux (34/51, 67%) and to swallowing difficulties. These difficulties often disappeared with ageing and most of them were later described as having a very good appetite (table 2).
The majority of patients (49/55, 89%) had recurrent and severe respiratory infections requiring numerous hospitalisations, and recurrent ENT (pharyngitis, otitis, sinusitis) and urinary tract infections. These infections and their complications were the primary cause of premature death (table 2). We also noted significant obstructive sleep apnoea (16/49, 33%), leading to adenoidectomy or tonsillectomy and sometimes to non-invasive mechanical ventilation.
Three patients (F41P2, F27P1, F27P3) suffered from pulmonary hypertension, which led to premature death, unrelated to a severe pulmonary infection, in two of them at the age of 11 months and 3 years, respectively. Another (F12P1), with right ventricular hypertrophy, died at the age of 19 years. One patient had been operated on for an atrial septum defect, one had a situs inversus and another had Bouveret tachycardia.
Urogenital anomalies were present in 43% of the patients (23/53) and included unilateral or bilateral cryptorchidism in 26% (14/53), micropenis in 9% (5/53), bilateral ureteral dilation in 9% (5/53) and bladder dilation or hypertrophy in three patients (table 2).
Gynecomastia was observed in three patients.
Discussion
Two large series of patients harbouring a MECP2 duplication have been reported to date,10 12 including 129 and 56 affected patients, respectively. In addition, we previously reported the MRI data of 30 affected males and Bauer et al reported the immunological phenotype of 27 affected patients.27 Our present series describes the demographic and clinical features of 59 French male patients with a focus on the morphological and neurodevelopmental features in order to improve the phenotypical characterisation of this syndrome. However, given that this disorder can be easily diagnosed using Array-CGH, which is commonly requested in the diagnosis of ID, the results presented in this study will be of value for the follow-up of patients.
There was a wide age range, from 2 to 48 years, the latter being the oldest male patient diagnosed to date with MECP2 duplication syndrome to our knowledge, Friez et al having reported a man aged 46 years.28 Among the whole French series of 86 male patients with an intraC MECP2 duplication, 27% died before the age of 25 years (11 months to 24 years, mean age: 9.5 years), from respiratory infections for 73% of them—which is similar to what has already been reported10 12—pulmonary hypertension for 18% and epilepsy for 9%. Like other authors, we were not able to demonstrate a clear correlation between the size of the duplication and the severity of the neurodevelopmental phenotype as the patients in our series with the smallest duplication did not have a milder neurological phenotype.15 22 29
We observed that the large majority of the patients were born at term and with normal birth parameters (online supplementary table 2). Few fetal anomalies were noted during pregnancies, including brain malformations, intrauterine growth retardation in two cases and bilateral hydronephrosis in two cases. In the same way, Fu et al reported four fetuses harbouring a MECP2 duplication and indicated that ventriculomegaly, hydrocephalus, agenesis of the corpus callosum, choroid plexus cysts, fetal growth retardation and hydronephrosis might be recurrent obstetrical ultrasound findings in fetuses with this disorder.30 We observed obstetrical complications in 12.5% of pregnancies, including polyhydramnios (7.8%), premature delivery threats (5.9%) or toxaemia (3.9%). Lim et al observed the same proportion of polyhydramnios and more hypertensive disorders (19%).12
We had previously described the OFC values in a smaller series of 30 patients and shown that there were nearly as many with microcephaly (13%) as with macrocephaly (17%).31 In the present series, we found that 7.5% of the patients were microcephalic and 17% were macrocephalic, which confirms that the OFC is not a key clinical feature in this syndrome (table 1). MECP2 duplication syndrome is classically described by a core phenotype that includes severe developmental delay with absent or very poor speech, epilepsy, respiratory infections and chronic constipation.7 9 Additionally, through a detailed analysis of the patients’ phenotype during consultations and photographs taken at different ages, our study showed other similarities emerging between the patients affected by this condition including distinctive facial features and dental anomalies including shape and eruption anomalies (table 1, figures 2 and 3). However, these signs alone are not sufficient to raise the diagnosis of MECP2 duplication clinically. We studied the evolution of the facial features with ageing and noted that they became coarser and that the syndrome was more difficult to recognise in adulthood, as usual in syndromology.32
In the literature, these patients are frequently reported to be ataxic.7 10 We noted in the present study that they mainly had a flexion attitude of the knees with a broad-based stance, mainly due to both hypotonia and spasticity, which led to a peculiar and unsteady gait (figure 4A). Another particularity was a decreased sensitivity to pain (29/37), which was also reported by Lim et al in 50% of their series.12
We also noticed frequent vasomotor problems, and a livedo of the limbs. Some of these signs had already been reported individually, like in the patient reported by Clayton-Smith et al who was described with a thin appearing skin and cold red feet with poor circulation.7 The association of decreased sensitivity to pain, chronic constipation, vasomotor troubles and unsteady gait with an abnormal posture could possibly result from an abnormal development of the proprioceptive and nociceptive pathways. Most of the patients developed kyphoscoliosis, which is important to take into account in their clinical care in order to start physiotherapy early in life and prescribe an orthopaedic corset when necessary. It seems important to note that some of these clinical features, including vasomotor troubles, small cold hands and feet, decreased response to pain, the stereotypic movements, bruxism and gait abnormalities are also supportive criteria of Rett syndrome. However, in MECP2 duplication syndrome, the development is abnormal from the outset and we do not observe frequently a deceleration of the OFC or breathing disturbances when awake.33
In our series, among the 32/59 patients with epilepsy, 62% were drug-resistant, compared with 76% in the series of Lim et al.12 Multiple different seizure types (table 2) and different EEG patterns (data not shown) were observed, which is consistent with other reports.24 34 35 Peters et al previously described that among 17 males with MECP2 duplication syndrome, 7 (41%) exhibited regression in motor skills, which coincided with seizure onset in 6 of them, compared with 12/31 (39%) patients in the present series.36 We observed very frequent bruxism and hand stereotypies as did other authors.12 24 29Compared with typical Rett syndrome, in which the onset of stereotypies mostly precedes or coincides with the loss of purposeful hand movements between ages 1 and 4 years (stage II) and tends to decrease after age 10 years,37 38 stereotypies in MECP2 duplication patients tend to appear during school age and will persist, in a variable way, throughout life.
Although some MECP2 duplication patients have been reported as having autistic behaviour,8 29 we observed that most of them had good social interactions, although we could not confirm this clinical impression with standardised diagnostic tool since our series was partly retrospective. In the same way, Xi et al did not find any MECP2 duplication in a series of 82 autistics males, suggesting that this disorder is not frequent in patients with autism.39 Conversely, hearing loss is not often reported in the literature in MECP2 duplication patients. Since we found hearing impairment in 7/23 patients in our cohort, this sensory impairment must be systematically assessed to avoid worsening of communication skills.
Digestive problems, ranging from chronic constipation to CIPO, are well-known features in MECP2 duplication patients. Lim et al noted that chronic constipation was present in 80% of the subjects in their series.12 This is consistent with our findings (78%), but seems to be more frequent than in previous reports (table 3).10 11 Several hypotheses were raised on the origin of the chronic constipation. Clayton-Smith et al attributed this to the FLNA gene, often involved in the duplication, and supported by the fact that point mutations in this gene were found in X linked families with pseudo-obstruction.7 40 41 Fernandes et al, on the other hand, hypothesised that this feature may be linked to the duplication of L1CAM, which is expressed in the enteric nervous system, plays a key role in its development during embryogenesis and has been found to be mutated in some patients with either X linked hydrocephalus or acrocallosal syndrome and Hirschsprung disease.42–44 In our series, among 39 patients with chronic constipation and for whom molecular data were available, we found that the duplication included FLNA and L1CAM in 25 cases, only L1CAM or only FLNA in 9 and 4 cases respectively, and none of them in 1 case. Therefore, in addition to the frequent constipation rate described in patients with severe neurological handicap, we may speculate whether increased dosage of both FLNA and L1CAM genes could be responsible for the increased risk to develop a severe constipation in these patients.
Comparison of the clinical data from the present study and those reported by Van Esch et al and Lim et al 10–12
Recurrent and severe infections were a significant health issue for 89% of MECP2 duplication patients, as in the study by Lim et al, who found recurrent respiratory infections in 78% of affected males, three-quarters of them requiring hospital admissions within the first 2 years of life for supportive care and interventional procedures (mechanical ventilation, intravenous antibiotics, tracheostomy).12 Van Esch, in a review of 129 affected males, reported 70% of recurrent infections, especially of the respiratory tract.10 These results raise again the question of the use of a long-term prophylactic antibiotic therapy in these males. It has been shown that increased susceptibility to infections in MECP2 duplication syndrome is associated with defects in the interferon-γ-mediated TH1 response,45 IgA/IgG2 deficiency, low antibody titres against pneumococci and elevated acute-phase responses. Some authors suggest prophylactic IgG substitution in MECP2 duplication patients with low IgA/IgG2 rates.27
Three patients in our series suffered from pulmonary hypertension (PH), being lethal for two of them. Two other unrelated MECP2 duplication patients who had lethal pulmonary hypertension, have been reported (rarechromosome.org—Xq28 duplication; http://www.goldcoastbulletin.com.au/news/gold-coast/gold-coast-family-mourns-11monthold-after-mystery-heart-attack/news-story/4d1f28630e5b6780359721cad0e41988). Interestingly, Cronk et al showed that Influenza A infected MeCP2Tg3 mice, overexpressing MeCP2 at levels threefold to fivefold higher than normal, experienced PH and arterial narrowing, which contributed to infection-related mortality.46 In our series, we speculate that PH might be the cause of the sudden and unexplained death of some patients, especially as MECP2 duplication patients do not usually undergo regular cardiac evaluation. We believe that these findings are important enough to introduce a cardiac assessment in these patients from the first year of life and then regularly, especially in patients who experienced several pulmonary infections.
Urogenital anomalies, in particular cryptorchidism and micropenis, were more frequent (26%) than in the literature,8 23 44 47 48 and gynecomastia was present in three patients, suggesting the possible existence of hypogonadism in this syndrome.
In conclusion, we report on the clinical and morphological features of 59 affected males with interstitial MECP2 duplication in order to expand the clinical phenotype of MECP2 duplication syndrome. Most of the clinical findings were consistent with previous data and we demonstrated that these patients shared common facial features that tend to evolve with age. In addition, we described commonly associated morphological and neuro-orthopaedic features not reported to date, such as a trunk and knees-flexion habitus leading to an ataxic-like gait. Pulmonary hypertension appeared as a cause of early death in these patients, which suggests that patients may benefit from regular cardiac assessment.
Acknowledgments
The authors are deeply grateful to the patients and their parents for their participation in this study. The authors would like to thank the association Xtraordinaire for their financial support of the project, and the French Ministry of Health (PHRC RMLX 2008, Pr V. Des Portes) for its support. The authors would like to thank the AChro-Puce network for its help in collecting cytogenetic data. The authors would also like to thank Mr James Garfield for his helpful review of this article.
References
Footnotes
Contributors MM wrote the paper, MN, CD, CP, JA, JPB, SCB, TB, MT, SDS, SD, AM, CB, CRT, GL, MR, CM, JT, MBD, DS, PC, ALM, NM, CC, VS, VM, SR, BK, ACT, VK, SS, AV and RAP analysed the cytogenetic and molecular data. LF, JA, RT, FP, LP, ML, SJ, CS, CF, FL, OBT, AD, BI, JV, BL, LL, JMC, GP, AT, AG, VCD, MR, BE, GR, HJ, LB, CB, LP, PSJ, KD, FD, DL, NP, SS, CTR, JMP, CG, CVD, BDB, NBB, AA, KM, CLC, DH, JLA, GL, MLPL, MR, BGD, MW, MC, HVE and DMC are the referring clinicians. LG interpreted brain MRI of many patients of the cohort, VD and NP respectively directed and organised the PHRC RMLX, SEC collected the clinical and molecular data and the photographs during consultations (for 25 patients) or by contacting the clinicians and the labs, and supervised this work.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval The ethics committee “Comité de Protection des Personnes SUD-EST II” delivered a favourable opinion (22 September 2010) with regard to the realisation of the XLID Research Project 2008-2016 (RMLX).
Provenance and peer review Not commissioned; externally peer reviewed.