Article Text

Abstract
Background Autosomal-recessive non-syndromic intellectual disability (ARNS-ID) is an aetiologically heterogeneous disorder. Although little is known about the function of human cereblon (CRBN), its relationship to mild cognitive deficits suggests that it is involved in the basic processes of human memory and learning.
Objectives We aim to identify the genetic cause of intellectual disability and self-mutilation in a consanguineous Saudi family with five affected members.
Methods Clinical whole-exome sequencing was performed on the proband patient, and Sanger sequencing was done to validate and confirm segregation in other family members.
Results A missense variant (c. 1171T>C) in the CRBN gene was identified in five individuals with severe intellectual disability (ID) in a consanguineous Saudi family. The homozygous variant was co-segregating in the family with the phenotype of severe ID, seizures and self-mutilating behaviour. The missense mutation (p.C391R) reported here results in the replacement of a conserved cysteine residue by an arginine in the CULT (cereblon domain of unknown activity, binding cellular ligands and thalidomide) domain of CRBN, which contains a zinc-binding site.
Conclusions These findings thus contribute to a growing list of ID disorders caused by CRBN mutations, broaden the spectrum of phenotypes attributable to ARNS-ID and provide new insight into genotype–phenotype correlations between CRBN mutations and the aetiology of ARNS-ID.
- ARNS-ID
- CRBN gene
- self mutiliation
- ubiquitination site
- intellectual disability
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Intellectual disability (ID) is primarily a neurodevelopmental disorder that affects 1–3% of the general population.1 It is characterised by an overall IQ of ≤70, associated with functional deficits in adaptive behaviour, social skills and communication, and has an onset before the age of 18 years.2 More than 40 genes linked to autosomal-recessive non-syndromic intellectual disability (ARNS-ID) have been identified, eight of which (PRSS12, CRBN, CC2D1A, GRIK2, TUSC3, TRAPPC9, ZC3H14, MED23) have been found by examining consanguineous families with members affected by ARNS-ID.3 ,4
Cereblon (CRBN) is located at locus 3p26.3 and is highly expressed in human tissues such as the hippocampus, cerebral cortex, cerebellum and striatum.5 ,6 CRBN is an ATP-dependent LON protease that contains a LON (DNA-binding ATP-dependent protease La) domain in addition to a CULT (CRBN domain of unknown activity, binding cellular ligands and thalidomide) domain, where all crucial residues of the thalidomide-binding site reside, and are conserved.7 CRBN forms an E3 ubiquitin ligase complex together with damaged DNA-binding protein 1, cullin-4A/B and regulator of cullins 1 and exists as a CRL4CRBN complex.8 The CRL4CRBN complex ubiquitylates a number of proteins through a mechanism that is not fully understood. However, CRBN is known to play a role in memory and learning by regulating the assembly and neuronal surface expression of large-conductance calcium-activated potassium (BKCa) channels. These ion channels are known to regulate membrane excitation, and mutations of ion channels often cause severe neurological disorders including seizures and epilepsy.6 ,9
To date, only one CRBN mutation (p.R419X) has been reported in the literature within ARNS-ID families.10 Here, we report a missense mutation in the CRBN gene that segregates with cognitive deficits in five affected members of a large consanguineous Saudi family. Whole-exome sequencing (WES) was carried out to establish the genetic cause of cognitive impairment in this family. Affected individuals exhibited severe ID, developmental delay, seizures and self-mutilating behaviour, thus expanding the spectrum of symptoms associated with ARNS-ID.
Methods
Subjects
A large consanguineous family from Saudi Arabia with five affected members (including three males and two females) was recruited in 2008 to the Department of Pediatric Genetics, King Abdulaziz Medical City, Ministry of National Guard Health Affairs, Riyadh, Saudi Arabia. The proband patient (IV.3, figure 1A) is now 25 years old, who is affected with intractable seizures, self-mutilating behaviour with severe ID, developmental delay and speech delay. However, his physical examination identified no dysmorphic or coarse features. The mother had all five kids born by normal vaginal delivery. The affected individuals started having self-mutilating behaviour around the age of 4 years. The proband patient (IV.3) is now on three anticonvulsants, besides the vagal nerve stimulator that was implanted to control his intractable seizures. On further investigation, it was revealed that patient IV.3 has four sisters, two of whom (IV.1 and IV.5, figure 1A) are affected by severe ID and self-mutilating behaviour. The two sisters (IV.1 and IV.5) started having seizures at 8 months and 2 years of ages, which were controlled by one anticonvulsant (valproic acid). Fortunately, they were off medication by 13 years of age. The younger sister (IV.5) has pelvic kidneys but with normal functions. She has severe self-mutilation to the extent that she even uses a nail clipper to cut her soles. Consanguinity at first cousin level was observed in the family (figure 1A), and two nephews (V.1 and V.2) were also suspected to be affected by ID as they were also having developmental delay. The nephews (V.1 and V.2) are now 4 and 3 years old, respectively. They have severe attention-deficit hyperactivity disorder and were only able to walk at 2 years of age. The older nephew (V.2) has seizures and no speech, whereas the younger one is more attentive compared with the older one and has only febrile seizures.
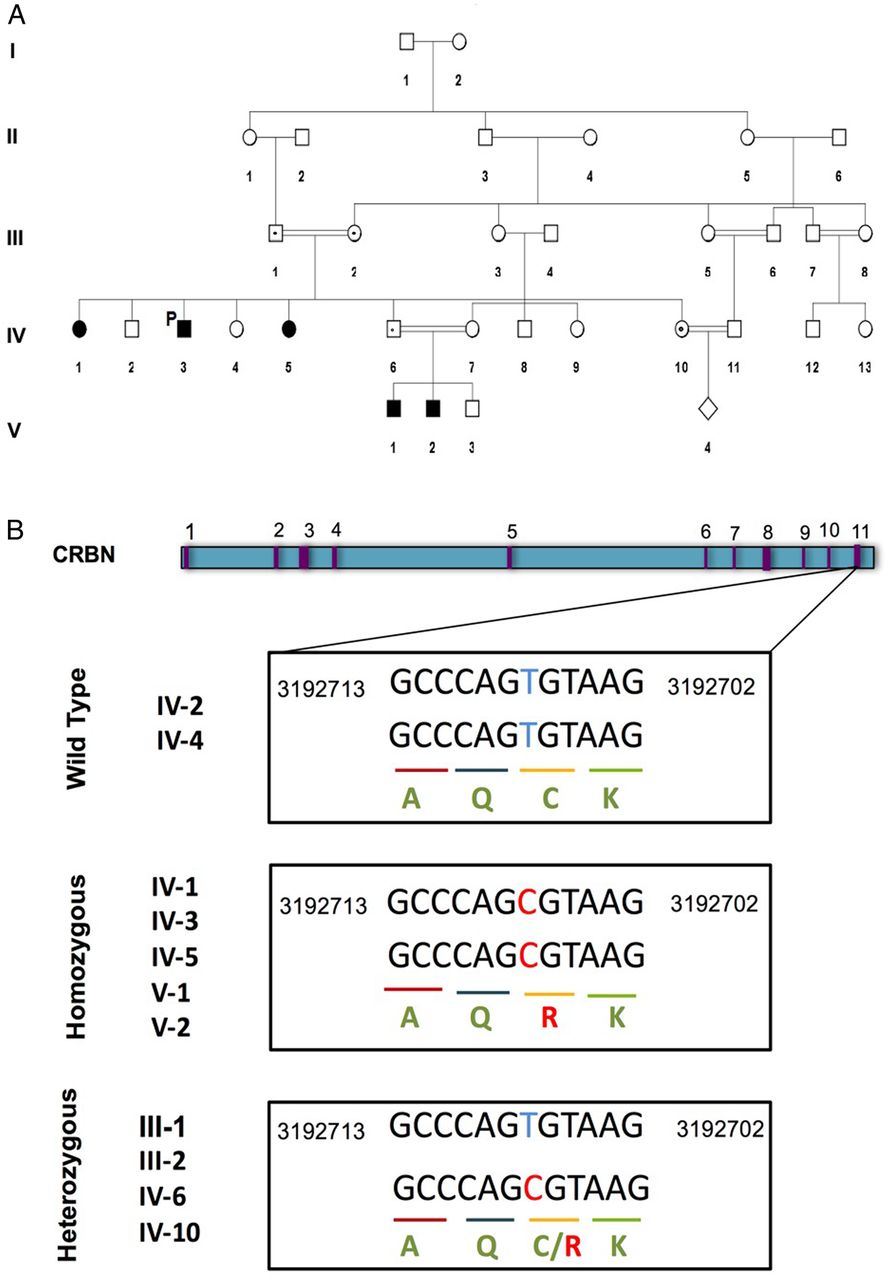
A family pedigree of a five-generation consanguineous family with autosomal-recessive non-syndromic intellectual disability (A). Patient IV.3 is the proband patient indicated as P (proband). These individuals, denoted by shaded symbols, are homozygous for a missense mutation in the CRBN gene. Family members heterozygous for this mutation are indicated with a black dot. (B). The genomic structure of the CRBN gene, with exons indicated in purple. The boxes underneath show exon 11 sequence containing the missense c.1171T>C mutation in homozygous and heterozygous individuals. It is aligned with a reference sequence from Human Genome GRCh37/hg 19 assembly. CRBN, cereblon.
The brain MRI and CT scan of the proband patient (IV.3) were unremarkable. His EEG showed slow background in the range of delta, and sometimes intermixed with theta activities. There were frequent spikes and waves seen over the left hemisphere with phase reversal at F3. The patient's (IV.3) biochemistry test profile revealed abnormal calcium and uric acid levels. The calcium was measured to be 1.98 mmol/L (ref range: 2.10–2.55 mmol/L) and uric acid was 164 µmol/L (ref range: 210–420 µmol/L), which were quite low. However, other metabolites—including sodium, potassium and magnesium—were within normal ranges.
Written consents were taken from the participants, or from their parents in the cases where affected individuals were under 18 years of age. The Institutional Ethical Review Board at King Abdullah International Medical Research Centre, Riyadh, approved the consent forms. Peripheral blood samples were collected in EDTA tubes from the proband, his parents and other affected siblings only after obtaining the informed consent. DNA was extracted using Puregene Extraction Kit (Qiagen, Germany).
Whole-exome sequencing
WES and interpretation of results were performed by Baylor College of Medicine, Medical Genetics Laboratory (http://www.bcm.edu/research/medical-genetics-labs/). WES was performed on DNA (extracted using Puregene Extraction Kit) from patient IV.3 (the proband, figure 1A). WES also included mitochondrial genome screening.
Data analysis and interpretation
The output data of Illumina HiSeq were converted from a bcl file to FastQ file by the Illumina CASAVA V.1.8 software and were mapped by the Burrows Wheeler Aligner program.11 The variant calls were performed using Atlas-SNP and Atlas-indel developed in-house by Baylor College of Medicine Human Genome Sequencing Center. The variants were interpreted according to the American College of Medical Genetics and Genomics guidelines and patient phenotype.12
Sanger sequencing
Segregation of potential mutations was validated using Sanger sequencing by Baylor College of Medicine. Sanger sequencing was performed on DNA from individuals of two generations to confirm the variant detected by WES in the IV.3 proband. Chromatograms were examined using Sequencing analysis software V.5.3 (Applied Biosystems, USA) and ClustaIW2 website. In silico analysis was performed using PolyPhen-2 and Scale-Invariant Feature Transform (SIFT).13
Results
WES of the proband patient (IV.3) revealed eight variants of unknown clinical significance associated with diseases related to the clinical phenotypes of our patient. Out of the eight genes, only CRBN was present as a homozygous variant (homozygous c. 1171T>C substitution). The presence of the mutation was validated with Sanger sequencing (as shown in figure 1B), which confirmed that the symptomatic sisters (IV.1 and IV.5) and nephews (V.1 and V.2) are homozygous for the c. 1171T>C mutation. The parents (III.1 and III.2), a sister (IV.10) and a brother (IV.6) of the proband patient were heterozygous for c.1171T>C, while one sister (IV.2) and a brother (IV.4) were negative (figure 1A,B).
The variant identified is located on exon 11 of the CRBN gene, which causes a cysteine residue to be changed into an arginine residue at position 391 (p.Cys391Arg) in the CULT domain of CRBN. This mutation is predicted to be deleterious by in silico analysis using software such as SIFT and PolyPhen-2 with values of 0 and 0.800, respectively. The cysteine residue at position 391 is conserved among vertebrates, as depicted from the UCSC genome browser (Chr 3: 3192707; GRCh37/hg 19 assembly), which suggests that this position might be involved in many aspects of the structure and function of CRBN including its target specificity.
Discussion
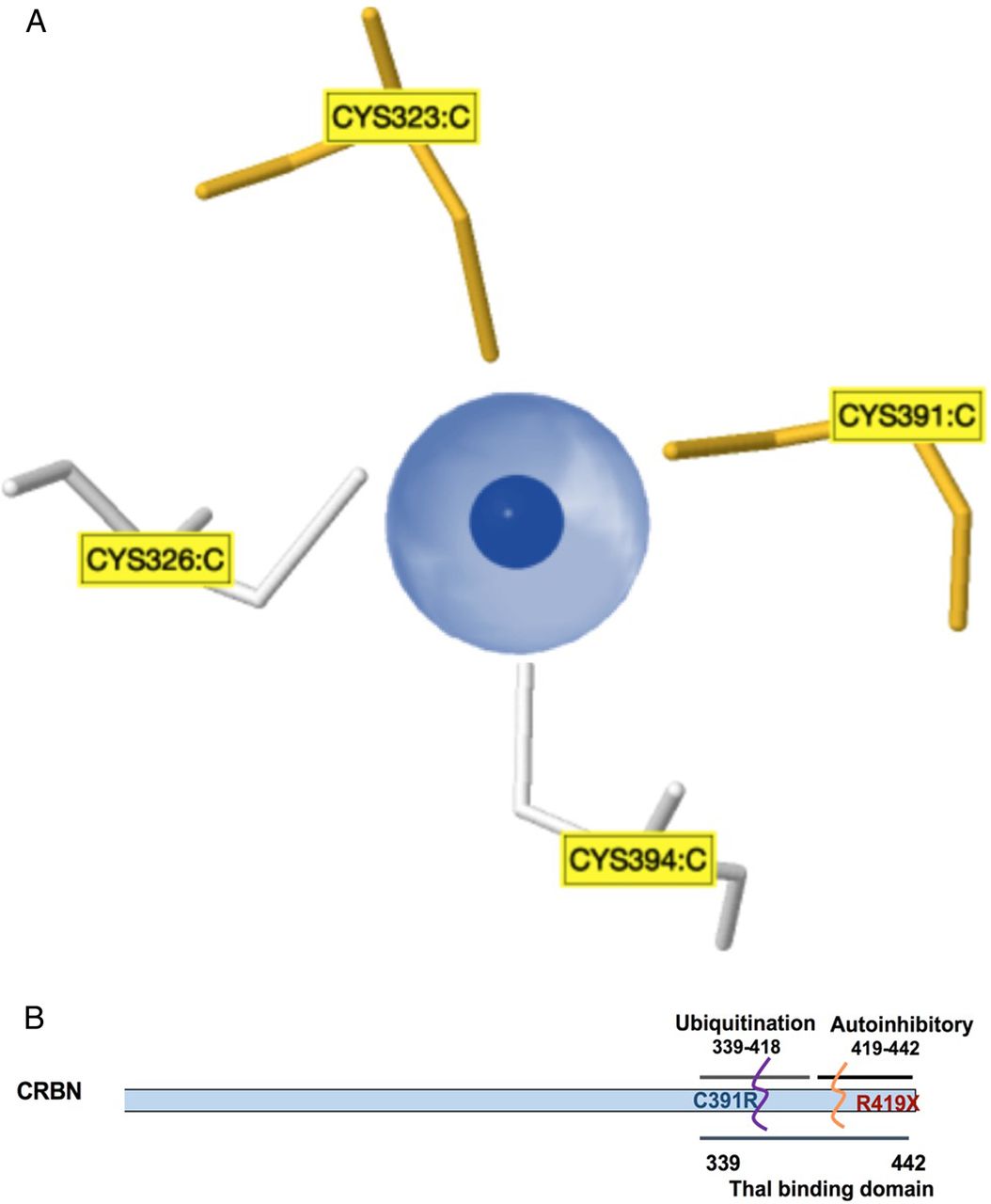
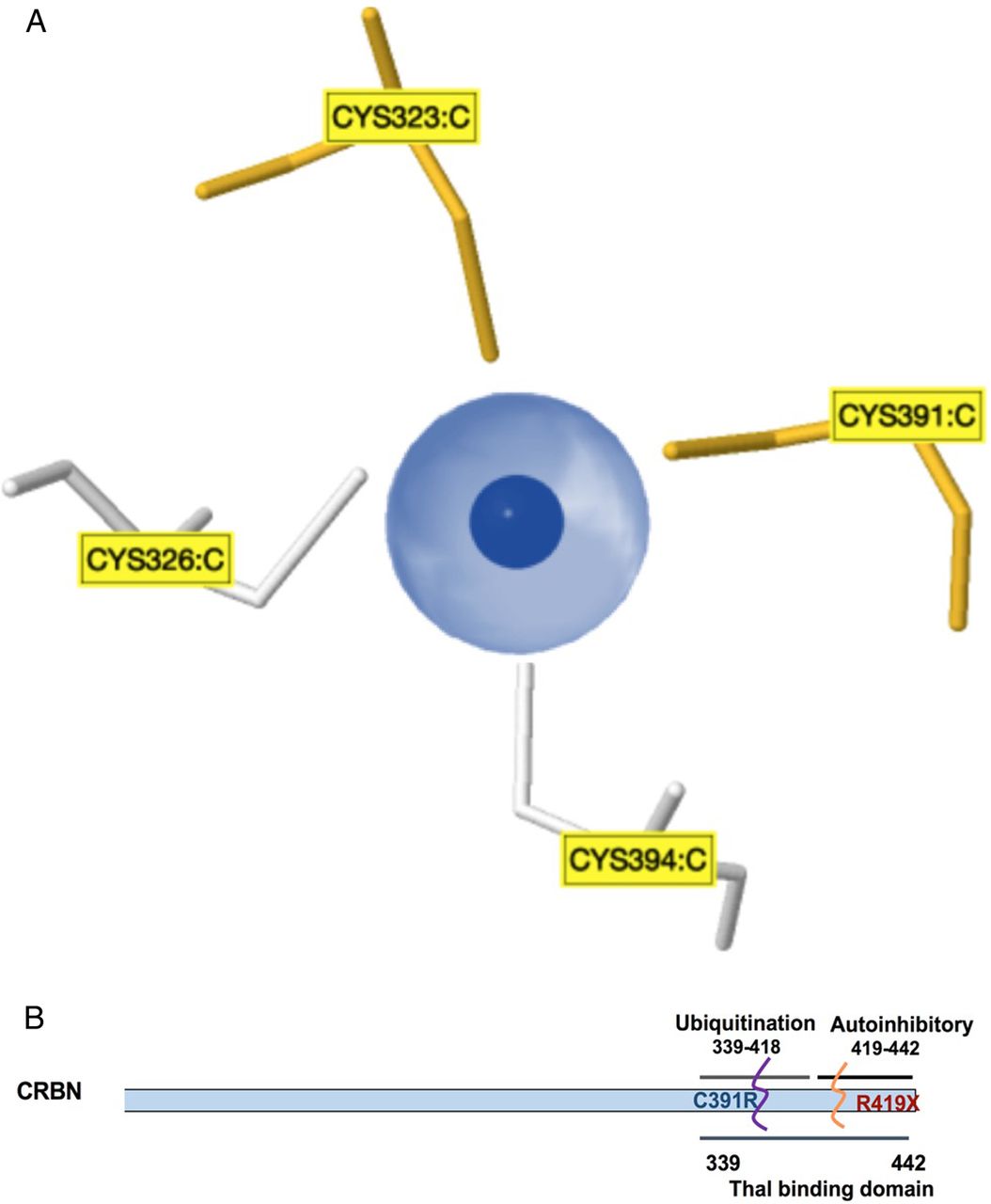
The identification of the self-mutilating behaviour with severe ID in this Saudi family is an additional feature that broadens the phenotypic spectrum associated with CRBN gene mutations linked to ARNS-ID. At position 391 of the CRBN protein, the cysteine binds to zinc (indicated as dark blue in figure 2A) and to three other cysteines at positions 323, 326 and 394 to maintain the tetrahedron structure of CRBN proteins (see figure 2A). It is known that cysteine residues display high affinity towards zinc ions, and these resulting Zn2+-cysteine complexes are essential mediators of protein structure, catalysis and regulation. The sulfur atom of cysteine transfers most of the charge to the zinc ligands and prevents the zinc to act as Lewis acid, thereby contributing to complex stability.14 Earlier, Valle and Auld15 proposed that these tetradentate zinc complexes have a high stability constant to ensure both the local and overall structural stability of the protein. The severe ID, intractable seizures and self-mutilating behaviour observed in the proband and other affected family members might have resulted from a loss of structural stability of the CRBN protein when a cysteine residue at a conserved site is replaced by an arginine residue. Moreover, Xu et al16 have proposed that amino acid residues, 339–418, in the CRBN protein form a major ubiquitylation domain as shown in figure 2B. We therefore deduce that the mutation p.Cys391Arg reported in this Saudi family disrupts this ubiquitylation site. Although little is known about the function of human CRBN, the association of this protein with cognitive deficits suggests that it is involved in fundamental processes of human memory and learning.6
{kind=link}
{kind=link}
Model and structure of cereblon (CRBN) (A). Zinc pocket interaction: a three-dimensional view of the Zn2+-interaction site in the CRBN protein. These zinc-binding sites do not contain a regular pattern of spacer length between the protein and the zinc ligands, and the ligands can be located on a flexible loop rather than in a rigid secondary structure. (B). Schematic representation of the CRBN gene showing C-terminal domains, with a suggested location for the p.Cys319Arg mutation at the ubiquitylation site that overlaps with the thalomide (Thal)-binding site.
The direct involvement of CRBN in the assembly and surface expression of large-conductance Ca2+-activated K+ channels (BKCa) has already been identified,17 although the mechanism by which CRBN interacts with the channel is not known. The large conductance, BKCa channels are expressed in brain neurons where they play an important role in regulating the duration of an action potential, firing frequency and neurotransmitter release.18 BKCa channels are governed by ubiquitylation, which controls the cell-surface expression and activity.19 Protein ubiquitination has been known as a universal quality control mechanism for the regulation of protein trafficking and turnover, and has been implicated in the dynamic control of diverse cellular processes.19
Usually, BKCa channels are targeted by the E3 ubiquitin ligase CRL4ACRBN for polyubiquitination and are retained in the endoplasmic reticulum (ER).19 Furthermore, CRBN co-localises with BKCa channels in brain neurons and regulates their surface expression.8 The CRBN gene is highly expressed in the hippocampus, consistent with its role in the pathogenesis of limbic seizures.9 Compelling evidence suggests that BKCa channel loss-of-function mutations contribute to neuronal hyperexcitability that leads to enhanced seizure susceptibility,20 although the mechanistic link between increased BKCa expression and ARNS-ID remains undefined. Kyle and Braun21 explain that CRBN, as a substrate of the CRL4ACRBN complex, interacts with BKCa in the central nervous system (CNS), leading to the ubiquitylation of the BKCa α subunit and retention of the channels in the ER. Any changes that affect the CRL4ACRBN complex could prevent ubiquitylation of the BKCa channels, which increases trafficking of these channels to neuronal cell membrane and increases the incidence of seizures and epilepsy in mice.9 Hence, the elevated BKCa channel expression in the CNS is closely linked with epilepsy, strongly suggesting that increased BKCa current density can lead to neurological disorders and possibly synaptic dysfunction/degeneration.
Ultimately, how the CRBN mutation reported in this study leads to ID with additional features at a mechanistic level remains unsolved and requires further investigation. Given that CRBN transcripts are widely expressed in tissues outside of the CNS, this regulatory paradigm may have broader functional implications. Thus, we hereby identify the genetic cause of ARNS-ID in five affected Saudi individuals and report a novel CRBN missense mutation that disrupts the protein's ubiquitylation site.
Acknowledgments
The authors thank the family for participating and supporting this study.
References
Footnotes
Contributors ASH and MAA contributed equally in writing the paper. SB prepared the initial draft of the manuscript. YAY assisted in developing figures, images and table for the manuscript. WE and SM supervised and supported the study.
Funding Ministry of National Guard Health Affairs, Riyadh, provided financial support.
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study was approved by the Institutional Review Board Committee at King Abdullah International Medical Research Centre, Riyadh.
Provenance and peer review Not commissioned; externally peer reviewed.