Article Text

Abstract
Background Early myoclonic encephalopathy (EME), a disease with a devastating prognosis, is characterised by neonatal onset of seizures and massive myoclonus accompanied by a continuous suppression-burst EEG pattern. Three genes are associated with EMEs that have metabolic features. Here, we report a pathogenic mutation of an ion channel as a cause of EME for the first time.
Methods Sequencing was performed for 214 patients with epileptic seizures using a gene panel with 109 genes that are known or suspected to cause epileptic seizures. Functional assessments were demonstrated by using electrophysiological experiments and immunostaining for mutant γ-aminobutyric acid-A (GABAA) receptor subunits in HEK293T cells.
Results We discovered a de novo heterozygous missense mutation (c.859A>C [p.Thr287Pro]) in the GABRB2-encoded β2 subunit of the GABAA receptor in an infant with EME. No GABRB2 mutations were found in three other EME cases or in 166 patients with infantile spasms. GABAA receptors bearing the mutant β2 subunit were poorly trafficked to the cell membrane and prevented γ2 subunits from trafficking to the cell surface. The peak amplitudes of currents from GABAA receptors containing only mutant β2 subunits were smaller than that of those from receptors containing only wild-type β2 subunits. The decrease in peak current amplitude (96.4% reduction) associated with the mutant GABAA receptor was greater than expected, based on the degree to which cell surface expression was reduced (66% reduction).
Conclusion This mutation has complex functional effects on GABAA receptors, including reduction of cell surface expression and attenuation of channel function, which would significantly perturb GABAergic inhibition in the brain.
- epileptic encephalopathy
- Epilepsy and seizures
- trafficking
- Cl<sup>-</sup> current
- GABA<sub>A</sub> receptor
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- epileptic encephalopathy
- Epilepsy and seizures
- trafficking
- Cl<sup>-</sup> current
- GABA<sub>A</sub> receptor
Introduction
Early myoclonic encephalopathy (EME) is an epileptic encephalopathy (EE) that, owing to its unique seizure phenotypes and EEG traces, is distinct from other early infantile EEs or Ohtahara syndrome (OS).1 EME is characterised by the onset of fragmentary erratic myoclonic seizures that are seen within the first 10 days of life and a suppression-burst (SB) pattern in EEGs during all sleep and wakefulness states that is precipitated by deep sleep. Over 50% of children with EME die within the first or second year of life.2 EME phenotypes are also seen mostly in metabolic disorders (eg, non-ketonic hyperglycaemia, amino and organic acidopathies, urea cycle disorders, mitochondrial disorders and pyridoxine or pyridoxal-5-phosphate disorders); thus, most EMEs are syndromic. In contrast, a minority of patients are ‘non-syndromic’, as they present with sporadic EME in the absence of metabolic disorders. The genetic aetiologies of non-syndromic EME are largely unknown.
To date, three genes (ERBB4 [MIM: 600543], SIK1 [MIM: 605705] and SLC25A22 [MIM: 609302]) have been associated with EME.3–5 ERBB4 and SIK1 are documented oncogenes, while SLC25A22 encodes a mitochondrial solute carrier. None of three genes, therefore, has obvious direct associations with neuronal excitability or inhibition. This is in contrast with findings that most EE-causing mutations are in proteins that are closely associated with excitatory or inhibitory synaptic functions, such as ion channels including the γ-aminobutyric acid (GABA)-A (GABAA) receptors.
GABAA receptors are ligand-gated chloride ion (Cl−) channels and function as pentamers. Though there are multiple subtypes of each subunit (ie, α1–6, β1–3, γ1–3, δ, ε, π, θ and ρ1–3), a combination of α1, β2 and γ2 subunits is the predominant form of GABAA receptor in the mammalian central nervous system. GABAA receptors play a cardinal role in controlling neural excitability in the central nervous system. Accordingly, mutations in genes encoding α1, β1, β3 and γ2 subunits (GABRA1, GABRB1, GABRB3 and GABRG2, respectively) have been found in EEs.6 ,7 However, no mutations of the β2 subunit have yet been reported in EEs.
Here, we report the discovery of a de novo heterozygous missense mutation in the GABRB2 gene that encodes the β2 subunit of the GABAA receptor. Our in vitro studies indicate that this mutation causes EME via disruption of GABAergic inhibition in the brain.
Materials and methods
Genetic analysis
Gene panel sequencing was performed in 214 patients with epileptic disorders using a customised HaloPlex Target Enrichment System (Agilent, Santa Clara, California, USA) for 109 genes that are known or suspected to cause epileptic seizures. The target coverage was 98.78% (see online supplementary table S1). Samples were sequenced on a MiSeq instrument (Illumina, San Diego, California, USA). SureCall software (Agilent) was used for mapping the hg19 and for trimming and aligning of fragments. The frequency of each variant was estimated in several publicly available databases, including 1000 Genomes, Exome Aggregation Consortium (ExAC) browser, human genetic variation database, ESP6500 and database SNP142. Pathogenicity of each variant was predicted using SIFT, PolyPhen2 hvar, Mutation Taster, CADD and PhyloP100way vertebrate using the ANNOVAR software. Variants with a frequency >0.005 were classified as polymorphic. Inheritance was confirmed by PCR-Sanger sequencing of genomic DNA from the parents. Follow-up sequencing was performed for all exons and the intron-exon boundaries of GABRB2 (RefSeq accession number NM_021911) in three patients with EME and 166 patients with infantile spasms (ISs) by PCR-Sanger sequencing.
supplementary table
Frequency of benign missense variants in the 10 regions of the β2-subunit.
GABAA receptor subunit cDNA constructs
The cDNAs encoding human α1, β2 and γ2S GABAA receptor subunit subtypes were subcloned into the expression vector pcDNA3.1(+). The GABRB2 (c.859A>C) subunit mutation was generated using the QuikChange Site-directed Mutagenesis kit (Stratagene, La Jolla, California, USA) and confirmed by DNA sequencing. γ2S, a short isoform of the γ2 subunit of GABAA receptor, was used in this study.
Measurement of surface β2 subunit expression using flow cytometry
Measurement of surface expression of GABAA receptor β2 subunits using flow cytometry has been described previously.8 ,9 Briefly, HEK293T cells were transfected using polyethylenimine reagent (40 kD, Polysciences) at a DNA:transfection reagent ratio of 1:2.5 and harvested 48 hours after transfection. To express wild-type (α1β2γ2s) and mutant (α1β2(T287P)γ2s) receptors, a total of 3 µg of subunit cDNAs were transfected at a ratio of 1:1:1 into 6 cm dishes for most experiments except for whole-cell recording. For mock or single subunit expression, empty pcDNA3.1 vector was added to make a final cDNA transfection amount to 3 μg. The transfected HEK293T cells were removed from the dishes by trypsinisation and then re-suspended in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline supplemented with 2% fetal bovine serum and 0.05% sodium azide). Following washes with FACS buffer and permeabilisation with Cytofix/cytoperm (BD Biosciences, California, USA) for 15 min, cells were incubated with mouse monoclonal anti-β2/3 antibody (1:200) for 2 hours and then incubated with fluorophore Alexa-647-conjugated goat anti-mouse secondary antibody (1:2000) for 1 hour at 4°C. Cells were then washed with FACS buffer and fixed with 2% paraformaldehyde. The acquired data were analysed using FlowJo 7.1 (Tree Star, Oregon, USA).
Immunocytochemistry
HEK293T cells expressing the wild-type α1β2γ2S or the mutant α1β2(Thr287Pro) γ2S receptors were fixed with 4% paraformaldehyde and immunostained with mouse monoclonal anti-β2/3 subunit antibody alone or co-stained with monoclonal anti-β2/3 subunit antibody and rabbit polyclonal anti-γ2 subunits. Rhodamine-conjugated mouse IgG alone or in combination with Alexa-488-conjugated rabbit IgG was used to visualise the wild-type or mutant subunits. The images were acquired using a LSM 510 invert confocal microscope with 63× objective.
Electrophysiology
Whole-cell recordings were obtained from HEK293T cells (HEK293T/17, ATCC RL-11268) that were cultured as monolayers in 35 mm dishes (Corning). For the wild-type (α1β2γ2s) and mutant (α1β2(T287P)γ2s) receptors, 0.3 μg cDNA of each α1, β (β2 or β2(T287P)) and γ2S subunit and 0.05 µg cDNA of enhanced green fluorescent protein (to identify transfected cells) were transfected using X-tremeGENE9 DNA Transfection Reagent (Roche Diagnostics, 1.5 μL/μg cDNA). Whole-cell recordings from lifted HEK293T cells were obtained at room temperature and the external solution was composed of 142 mM NaCl, 8 mM KCl, 10 mM D(+)-glucose, 10 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 6 mM MgCl2.6 H2O and 1 mM CaCl2 (pH 7.4, ∼326 mOsm). The internal solution consisted of 153 mM KCl, 10 mM HEPES, 5 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid) 2 mM Mg-ATP and 1 mM MgCl2.6H2O (pH 7.3, ∼300 mOsm). The Cl− reversal potential was near 0 mV and cells were voltage-clamped at −20 mV. GABA (1 mM) was applied for 4 s using a four-barrel square glass connected to a SF-77B Perfusion Fast-Step system (Warner Instruments, Connecticut, USA). Whole-cell currents were amplified and low-pass filtered at 2 kHz using an Axopatch 200B amplifier, digitised at 10 kHz using Digidata 1550 and saved using pCLAMP 10.4 (Axon Instruments). Data were analysed offline using Clampfit 10.4 (Axon Instruments).
Structural modelling
Pentameric GABAA receptor homology models were generated by combining human α1, β2 and γ2 structural models with the subunit arrangement β-α-β-α-γ2 implemented in the programme suite incorporated in SWISS-MODEL (http://swissmodel.expasy.org/SWISS-MODEL.html). Three-dimensional models of human GABAA receptor subunits were generated using the crystal structure of the Caenorhabditis elegans glutamate-gated chloride channel10 as a template (PDB: 3rhw) as previously described.11 We prepared the figures using Chimera 1.7.
Results
Patient with EME
The proband is a boy aged 1 year and 10 months. He was born at term to non-consanguineous parents as the result of a natural pregnancy. He had an appropriate body size for his gestational age and had no physical malformations. There was no history of epilepsy, developmental disorders or other neurological disorders in his parents, an elder sister, or other relatives. Immediately after birth, the boy presented with poor feeding, lethargy and weak crying. Subtle eyelid and limb myoclonus was also noted. He had generalised tonic clonic convulsions (GTCCs) lasting 5–10 min at the age of 2 months and was admitted to the children medical centre of Fukuoka University. Neurological examinations on admission identified drowsy consciousness, poor response to stimulation and hypotonicity. Laboratory results of blood and urine examinations, including evaluation of amino acids, organic acids or lipid metabolism, were all normal and a brain MRI was normal. However, an interictal EEG recording showed a SB pattern during wakefulness (figure 1A). Sleep EEG also displayed the SB pattern, of which the suppression components consisted of slow waves with approximately 150 µV amplitude continuing for 4–12 s, with an average of 6.5; the burst components indicated multiple focal lesions. During recording of sleep video EEG, we observed myoclonus in the eyelids, which corresponded to the bursts. During the EEG recording, intravenous injection of pyridoxine did not affect the clinical or EEG findings. Myoclonus in the eyelid and limbs was seen frequently every day thereafter and GTCC with apneic spells was seen approximately once a day. These clinical findings suggested a diagnosis of early onset EE, specifically EME. As possible causes of EME, we first considered endocrine disorders, such as hyperthyroidism, and metabolic disorders, such as non-ketotic hyperglycinemia, amino and organic acidopathies, urea cycle disorders, mitochondrial disorders, pyridoxine and pyridoxal-5-phosphate disorders, molybdenum cofactor deficiency, sulfite oxidase deficiency, Menkes syndrome, Zellweger syndrome and other disorders, because of the appearance of a normal brain structure in MRI analysis. General serum biochemistry tests, including ammonia, lactic acid, pyruvic acid and copper levels, were normal. Urine tests, including the qualitative reaction of sulfurous acid, were normal. Serum free T3, T4 and thyroid-stimulating hormone levels were in the normal range. Mass spectrometry and amino acid analyses for serum, urine and cerebrospinal fluid (CSF) showed normal concentrations of glycine in the serum and urine, low CSF:serum ratios and normal levels of taurine, cystatin, citrulline, ornithine, arginine and other amino acids. Acylcarnitine analysis by tandem mass spectrometry also did not identify any abnormalities. Examination of the ocular fundus, abdominal ultrasonography and cardiac ultrasound did not identify any abnormalities. Although we also screened for methamphetamine, benzodiazepine, cocaine, phencyclidine, opioids, cannabis, barbiturates and tricyclic antidepressants by simple urine examination, all of these tests were negative. These results excluded hyperthyroidism and metabolic disorders. Chromosome G banding analysis showed a normal 46, XY karyotype.

EEG showing proband. Monopolar EEG. Red bar: Eye jerk. (A) Suppression-burst (SB) pattern during sleep at the age of 2 months. (B) SB pattern during sleep at the age of 1 year and 11 months.
Although phenobarbital and levetiracetam treatments were initiated for seizure control, there were no remarkable improvements. Epileptic spasms forming a series without hypsarrhythmia emerged at the age of 3 months as the frequency of GTCC decreased. Frequent myoclonus in the boy's eyelids and limbs continued and the SB pattern on EEG remained at the age of 1 year and 11 months (figure 1B). Together with the fact that the seizures were not triggered by fever, a diagnosis of EME was made. His psychomotor delay was severe and at the time of this study the patient was bedridden and was being fed via a tube.
Mutation analysis
Gene panel sequencing was performed for the baby boy suffering from EME (coverage: 93.57%). This identified a heterozygous missense variant in exon 9 of GABRB2 (RefSeq accession number NM_021911: c.859A>C [p.Thr287Pro]), which fulfilled the condition of ‘moderate’ with regard to pathogenicity according to the American College of Medical Genetics and Genomics guidelines.12 This variant was absent in controls in population databases including the Exome sequencing project, 1000 Genomes project, ExAC and SNP142. Predictive algorithms of SIFT, Polyphen-2 and MutationTaster estimated the variant as deleterious and CADD and phyloP returned very high scores (24.3 and 7.911, respectively). PCR-Sanger sequencing also confirmed the c.859A>C variant in the patient (figure 2B). We investigated co-segregation of the variant with phenotypes among family members, namely his asymptomatic parents and an elder sister. None of them harboured the variant and hence this heterozygous missense variant was considered de novo (figure 2A, B). Since it was classified as a very strong pathogenic variant, we labelled this as a mutant genotype. The Thr287 residue is highly conserved among species (figure 2C and online supplementary figure S1). We sought pathogenic variants in GABRB2 of three additional patients with EME, including the GM13078 line purchased from Coriell Institute (Camden, New Jersey, USA); all cases were negative. Similar mutations in the same gene may exhibit different types of EEs. For example, missense mutations of KCNT1 are found in autosomal-dominant nocturnal frontal lobe epilepsy13 and in migrating partial seizures in infancy14 or IS,6 whereas missense mutations of STXBP1 are found in OS, IS or Dravet syndrome.15 ,16 Additionally, the Epi4K consortium (2013) described de novo mutations in over 200 genes in 149 and 115 individuals with IS and Lennox-Gastaut syndrome (LGS), respectively.6 Remarkably, four de novo mutations of GABRB3, which encodes the β3 subunit of GABAA receptors, were identified in a patient with IS and in three patients with LGS (two of these LGS cases evolved after an initial diagnosis of IS). To elucidate whether mutations in GABRB2 were shared between EME and IS, we also looked for changes at this locus in 166 individuals with IS. However, no mutations were found. In addition, the Epi4K consortium, which conducted massive exome sequencing, did not find GABRB2 mutations in 149 cases of IS.6 Overall, no mutations in GABRB2 were found in 315 cases of IS.
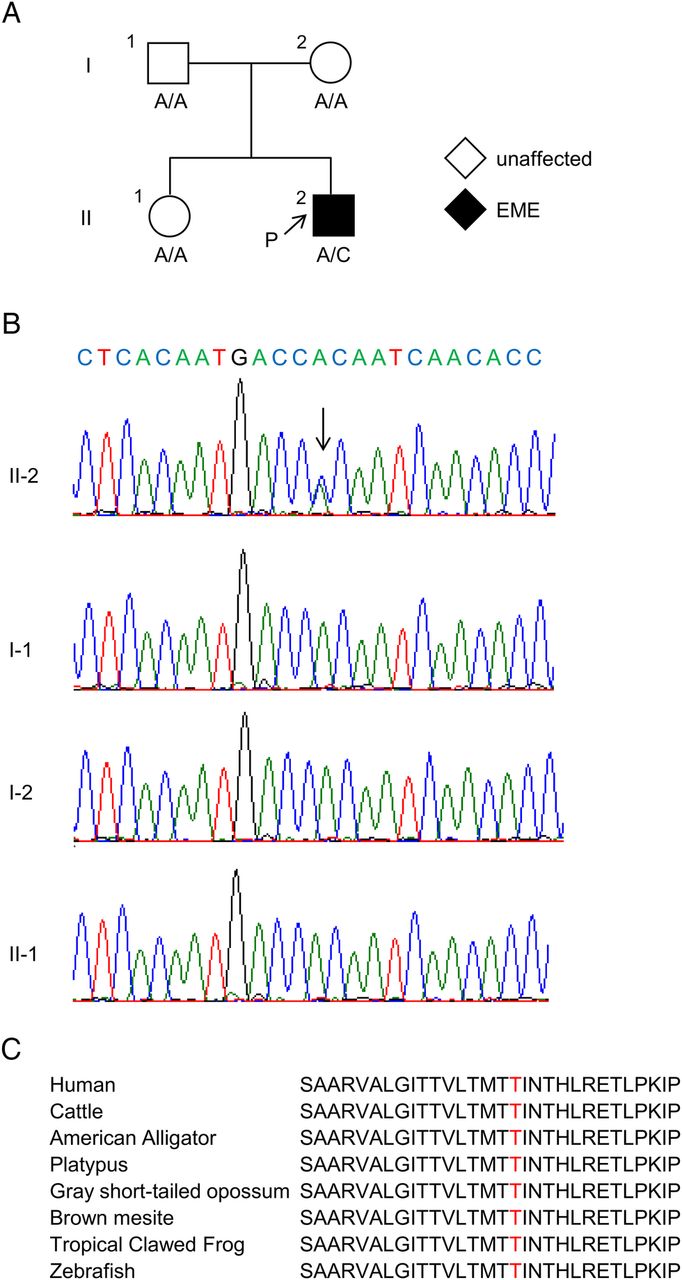
Pedigree and GABRB2 mutations are shown. (A) Pedigree and their genotypes. Arrow with ‘P’ indicates the proband of this family. Square, circle and black symbols indicate male, female and affected individual(s). Mutation (RefSeq accession number NM_021911: c.859A>C) was found only in the proband. (B) Chromatogram showing PCR-Sanger sequencing. Arrow indicates a C-to-A substitution. A missense mutation was found only in the proband of this family. (C) Interspecies conservation of the amino acid Thr287 is shown. Red indicates the amino acid at position 287. EME, early myoclonic encephalopathy.
supplementary figures
On the other hand, the ExAC browser indicates 297 missense variants at 95 distinct sites in the GABRB2 locus (accessed 28 March 2016). These findings suggested two possibilities: (1) GABRB2 mutations may not directly cause IS but may cause other phenotypes and (2) such mutations are extremely rare and may result in more severe, lethal or rare phenotypes. To understand the extent to which the missense mutation p.Thr287Pro is deleterious, we examined the distribution of 297 missense benign variants from the ExAC browser. These fell within the coding sequence (1539 bp) of GABRB2, which encodes 10 regions of signal peptides, four transmembrane domains (TM1 to TM4), the N/C-termini and three loops (see online supplementary table S2 and figure S3). Benign missense variants were not distributed evenly throughout the nucleotide sequence (Fisher's exact test, p value=0.0004998) (see online supplementary table S2). We then tested the distribution of benign variants in the amino acid sequence of each region. Two loop regions, between TM1–TM2 and TM3–TM4, appeared to accumulate benign variants more frequently (see online supplementary figure S3). The mutation rate was statistically significant at the loop between TM1 and TM2 (Fisher's exact test, p value=0.0468 and two-sample proportion test, p value=0, respectively). We then compared the fraction of benign variants in each of the above four regions with the fraction of 296 benign variants along the remaining 433 amino acids length. This revealed a statistically significant reduced accumulation of benign variants (Fisher's exact test, p value=0, respectively) in four regions (TM1, TM2 (where p.Thr287Pro is located), TM3 and the loop TM2–TM3). This result suggests that amino acid substitutions in TM2 are not tolerated and cause severe phenotypes such as EME.
Reduced total and surface protein expression of the Thr287Pro-mutant β2 subunit
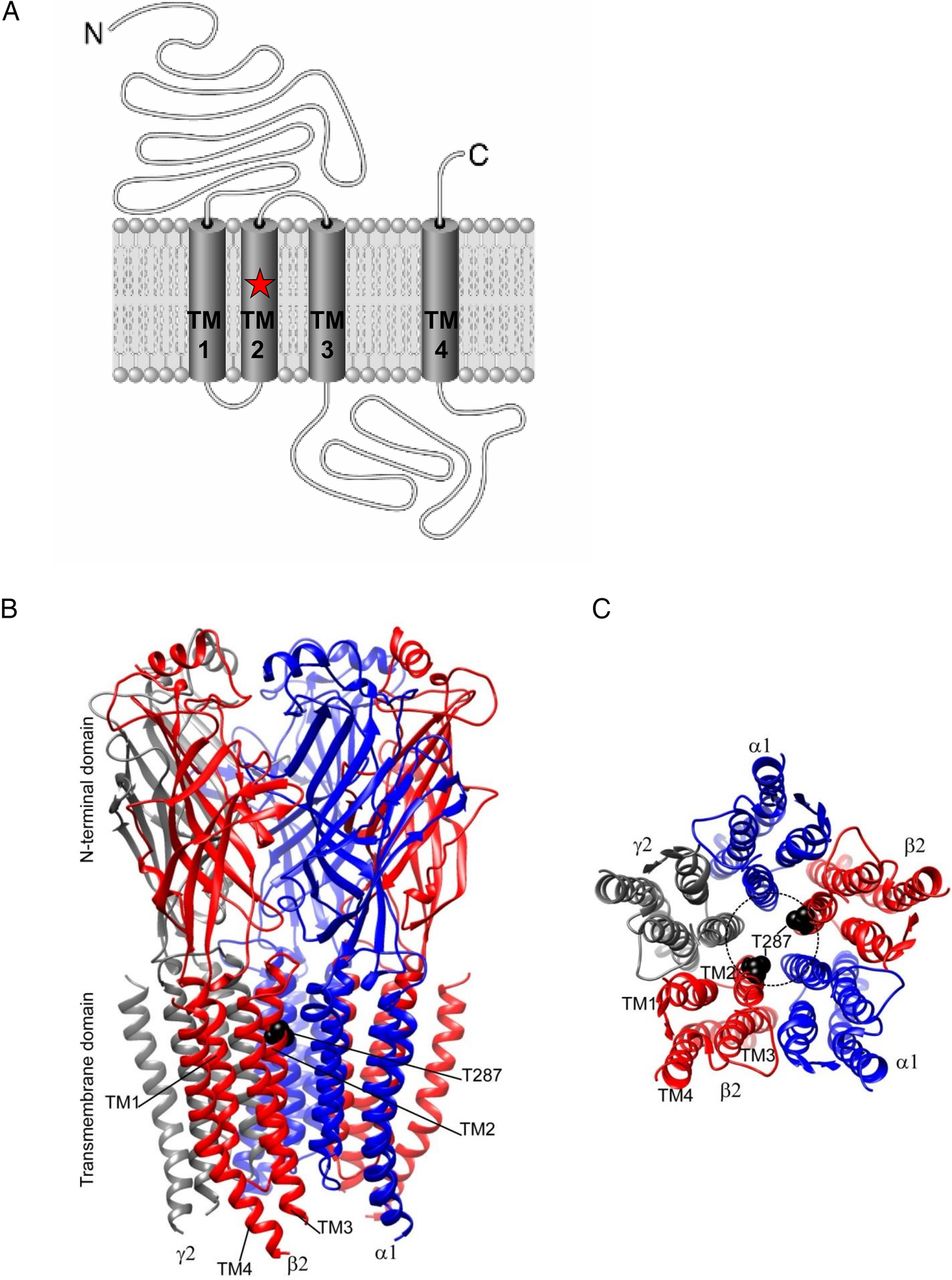
The β2 subunit of GABAA receptor consists of four transmembrane domains (TM1 to TM4) connected by loops and extracellular N and C termini (figure 3A). TM2, where the mutation resides, forms Cl− ion pore of GABAA receptors (figure 3B, C). The GABAA receptors in the central nervous system are pentamers consisting of two each of the α1 and β2 subunits and one γ2 subunit, which are encoded by GABRA1, GABRB2 and GABRG2, respectively (figure 3B). We asked whether the p.Thr287Pro mutation might impair Cl− ion channel function of the GABAA receptors, which might in turn hamper GABAergic neuronal inhibition. Furthermore, since we have demonstrated that trafficking deficiency is a major defect caused by GABRG2 mutations,9 ,17 we also examined trafficking of the mutant β2 (Thr287Pro) subunits. HEK293T cells were co-transfected with α1 and γ2 subunits and either wild-type β2 only, mixed wild-type β2 and the mutant β2 (Thr287Pro) subunits or the mutant β2 (Thr287Pro) subunits only (mut). Total and surface protein expression of the wild-type and mutant β2 subunits were then compared. We used sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblot to determine total β2 subunits (figure 4A). Compared with the total β2 subunits in the ‘wild type receptor only’ condition, both the mixed and the mutant β2 subunits (0.61±0.02 for mixed, 0.35±0.04 for mutant β2 (Thr287Pro) subunits vs 1 for wild type, n=4) were reduced when co-expressed with α1 and γ2 subunits (figure 4B). Reduced total β2 subunit expression could result in the reduced expression of surface β2 subunit. We next determined surface protein expression of the mutant β2 (Thr287Pro) subunit. We used the high-throughput flow cytometry to quantify the amount of surface wild-type or mutant β2 subunit when co-expressed with the α1 and γ2 subunits; this is because pre-assembled pentameric receptors are trafficked to the cell surface (figure 4C). Similar to the total protein expression levels, surface β2 subunits for both the mixed and mutant receptors were reduced (0.54±0.045 for mixed, 0.34±0.042 for mutant β2 (Thr287Pro) subunits vs 1 for wild type, n=4) (figure 4D).

p.Thr287Pro mutation in the β2 subunit and γ-aminobutyric acid-A (GABAA) receptor is shown. (A) Cartoon representation of the location of the p.Thr287Pro mutation of β2 subunit of the GABAA receptor. (B) Three-dimensional structural model of the GABAA receptor that is composed predominantly of two α1 (blue ribbons), two β2 (red ribbons) and one γ2 (grey ribbon) subunits in the mammalian central nervous system. β2 subunits have four transmembrane domains (TM1 to TM4). The GABRB2 de novo p.Thr287Pro mutation is mapped onto the β2 subunit in black at the second transmembrane domain (TM2). (C) Extracellular view of the transmembrane domain in a structural model of pentameric αβγ GABAA receptor (The N-terminal domain was removed for clarity) displaying the GABRB2 mutations (in black) on β (red ribbons) subunits. TM2 domains of five subunits form the Cl− ion pore (dashed black circle) and the p.Thr287Pro mutations are within the pore region.

Reduced expression and cell surface levels of mutant β2 (p.Thr287Pro) (mut) subunit. (A, B). HEK293T cells were co-transfected with α1 and γ2 subunits and wild-type β2 (wt, 1:1:1 cDNA ratio of α1: β2: γ2), mixed wt β2 and the mut subunits (mixed, 1:0.5:0.5:1 cDNA ratio of α1: β2: β2(Thr287Pro):γ2) or the mut (1:1:1 cDNA ratio of α1: β2(Thr287Pro): γ2) subunits. (A) Total lysates were analysed by sodium dodecyl sulfate polyacrylamide gel electrophoresis and western blot. The membranes were blotted with rabbit anti-β2 subunit antibody. (B) The total β2 subunit protein integrated density values (IDVs) were normalised to the wt β2 subunit in wt receptors. (C) The flow cytometry histograms depict surface expression levels of β2 subunits from HEK293T cells expressing wt, mixed or the mut α1β2γ2 receptors. Cell surface wt and mut subunits were stained with monoclonal anti-β2/β3 (BD17) antibody that was fluorescently conjugated with Alexa Fluor-647). (D) The relative fluorescence intensity of β2 subunit signals of the mut subunits were normalised to those obtained with wt β2 subunits in the wt receptors (in (B) and (D), **p<0.05, **p<0.01, ***p<0.001 vs wt). p Values were obtained by unpaired t-test.
Α1β2γ2 receptors containing mutant β2(Thr287Pro) subunits were retained inside cells
We previously demonstrated that the misfolded mutant GABAA receptor subunits were retained inside the endoplasmic reticulum (ER) and subsequently degraded without trafficking to the cell surface,18 thus resulting in reduced total and surface expression of the mutant subunits. To investigate if the mutant β2(T287P) subunits were also subject to ER retention and premature degradation, we determined the subcellular localisation of the mutant subunits. We first immunostained for either the wild-type or mutant β2(T287P) subunits in HEK293T cells co-expressing α1 and γ2 subunits. The wild-type β2 subunits were mainly present on the edges of cells, whereas the β2(T287P) subunits were mainly found close to the nuclei (figure 5A). Because the β2 subunit is required for α1β2 or α1β2γ2 subunit assembly, mutant β2 subunits may prevent trafficking of these partners to the cell surface. We thus performed co-staining experiments to determine whether mutant β2(T287P) subunits affected the localisation of the partnering wild-type subunits like α1 and γ2 subunits. This revealed that γ2 subunits were co-localised with β2 subunits and had similar subcellular presence as in the cells stained with β2 subunits alone (figure 5B).

Mutant β2 (p.Thr287Pro) subunits and their wild-type partner subunits were retained intracellularly. HEK293T cells expressing the α1 and γ2S subunits with the wild-type β2 or the mutant β2(p.Thr287Pro) subunits (1:1:1 cDNA ratio) were immunostained for anti-β2/3 subunits alone (A) or in combination with rabbit anti-γ2 subunits (B). The anti-β2/3 subunits were visualised with rhodamine-conjugated IgG, while the γ2 subunits were visualised with Alexa488-conjugated IgG. The images were acquired using a confocal microscope with a 63× objective based on our previous protocol.17
Α1β2γ2 receptors containing mutant β2(Thr287Pro) subunits have reduced peak current amplitude
Lower surface β2 subunit content may reduce receptor channel current amplitude, because this subunit is required for α1β2 or α1β2γ2 receptor assembly.19 We then measured the current amplitudes of α1β2γ2 receptors or α1β2(Thr287Pro)γ2 using patch-clamp whole-cell recording. We recorded currents evoked by 4-s applications of GABA from HEK293T cells co-transfected with α1 and γ2S subunits and either wild-type or mutant β2 subunits (figure 6A). The peak amplitude of the α1β2(Thr287Pro)γ2S receptor currents (0.26±0.10 nA, n=10) were smaller than that of wild-type α1β2γ2S receptor currents (7.3±0.51 nA, n=10) (figure 6B). This is consistent with the reduced surface expression of mutant β2(Thr287Pro). Furthermore, the decreased peak amplitude associated with the α1β2(Thr287Pro)γ2S receptor (96.4% reduction) was much more pronounced than would have been expected based on the degree to which surface expression was reduced (66% reduction), indicating that the mutation engenders Cl− channel dysfunction in addition to its deleterious effects on trafficking by reducing surface subunit expression (figure 4D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of mutant β2 (p.Thr287Pro) subunits reduces the peak current amplitudes of γ-aminobutyric acid-A (GABAA) channels. (A) Representative GABA current traces obtained following rapid application of 1 mM GABA for 4 s to lifted HEK293T cells voltage-clamped at −20 mV. The current traces from GABAA receptors containing the mutant β2(T287P) was compared with their respective wild-type (wt) α1β2γ2s current traces. (B) Bar graph shows the average peak current from cells expressing wt and mutant GABAA receptors. Values represent mean±SEM (n=10 patches). Statistical differences were determined using unpaired t-test; **** indicates p<0.0001 compared with the wt condition.
Discussion
Here we have shown for the first time that GABRB2 is associated with EME, which is one of the most severe EE forms encountered in the clinic. We believe that this is also the first report that a pathogenic mutation in a neuronal ion channel can cause EME.
We excluded the possibility of metabolic disorders and made a diagnosis of non-syndromic EME, although this case showed some overlap with OS both in EEG findings and in the seizure type, that is, generalised tonic-clonic seizure. However, in this case, video EEG revealed myoclonus during the burst stage of the SB pattern, hyposthenia in the suppression stage during sleep and movement of limbs in the suppression stage during awakeness. At the age of 1 year and 11 months, the predominant seizure type was myoclonus and the SB pattern was still present on EEG. Video EEG findings and continuous myoclonus, regardless of age, were distinctly different from OS.
In addition to GABRB2, three other genes (ERBB4, SLC25A22 and SIK1) are associated with non-metabolic EME. A genetic abnormality of ERBB4 was found in a patient with EME because of a de novo reciprocal translocation t(2;6)(q34;p25.3).3 This was not a single nucleotide variation (SNV) or indel and the patient had some dysmorphic features. A homozygous mutation of SLC25A22 was found in two siblings born to their consanguineous parents.5 Currently, SIK1 is the only gene of which heterozygous SNVs (including missense and nonsense mutations) have been identified as the cause of sporadic EME cases;4 several similar SIK1 mutations have also been found in IS and OS. All three genes have been implicated in cell metabolism and growth. To ask whether there were any relationships between GABRB2 and these three genes, we examined protein-protein interactions using the Search Tool for the Retrieval of Interacting Genes/Proteins (accessed 11 March 2016) (see online supplementary figure S2). However, we did not find any direct relationships between GABRB2 and ERBB4, SLC25A22 or SIK1, suggesting that there is significant heterogeneity for the aetiology of EME.
Another de novo heterozygous missense mutation (c.236T>C; p.Met79Thr) of GABRB2 was found in a sporadic case with mild intellectual disability and epilepsy.20 The patient was a 12-year-old girl who had her first seizure evoked by fever at the age of 9 months; this was followed by non-febrile GTCC in subsequent years. Her seizures responded to clobazam. Although her development slowed over the years, she was still able to attend regular school. Thus, the clinical symptoms of this patient harbouring the p.Met79Thr mutation were much milder and quite different from those of individuals with EME. The actual functional consequence of the p.Met79Thr mutation has not been evaluated. It is located in the N terminus of the β2 subunit, which is one of the regions prone to accumulation of benign variants (see online supplementary figure S3). However, the p.Thr287Pro mutation identified in the present case seems more deleterious than the p.Met79Thr variant, as it resides in TM2, which forms part of the Cl− pore. This likely explains the milder clinical phenotype associated with Met79Thr variants.
As follow-up study, we screened three patients with EME. This is too small a population to analyse the relationships between mutations and phenotypes. However, no GABRB2 mutations were found in any of the 315 cases with IS (this number includes cases in the Epi4K study)6, suggesting that the GABRB2 mutations may be more likely to be involved in the aetiology of EME than IS. According to the distribution of GABRB2 benign variants we have analysed, the chances that TM1, TM2, TM3 and TM2–TM3 loop have benign variants are significantly lower than other regions (Fisher's exact test, p value=0, respectively). These regions are considered ‘cold’ spots for benign variants. In addition, since GABRB2 is a small gene of 1539 nucleotides that encodes only 522 amino acids, the rate at which de novo variants emerge is low given that they arise randomly. Considering the low incidence of benign variants in such ‘cold’ regions, it is likely that most mutations in these regions would have a negative impact and thus cause rare, severe or even lethal phenotypes. This hypothesis is consistent with the fact that non-syndromic EME is one of rarest and most severe forms of EE.
In accordance with the severe phenotype of the present case, GABAA receptors bearing the mutated β2 subunit had several aberrant properties in vitro. For example, cell surface expression of p.Thr287Pro β2 subunits was significantly reduced compared with GABAA receptors with the wild-type subunit; this was most evident in the homozygous mutant state (figures 4D and 5A). We also showed that γ2 subunits and mutant β2(Thr287Pro) subunits were co-localised in cells, suggesting that they oligomerised as protein complexes (figure 5B). These findings can explain why α1β2γ2 receptors containing mutant β2(Thr287Pro) subunits are retained inside cells. Because of the β2 subunit's essential role in α1β2 or α1β2γ2 receptor assembly, it is likely that cell surface expression of all the wild-type partnering subunits, including α1 and γ2, is also reduced. GABAA receptors containing the mutant β2 subunits exhibited a much smaller peak current amplitude than those containing wild-type β2 subunits (figure 6). Interestingly, however, the reduction of peak current amplitude was not proportional to the reduction in cell surface expression. Indeed, reduction in peak current amplitude was much greater than would have been expected based on the levels of mutant present at the cell surface. This discrepancy suggests that, in addition to compromising subunit protein trafficking, the p.Thr287Pro mutation also functionally impairs the mutant GABAA receptors that do reach the surface. This impaired channel function may be due to a dominant negative effect of p.Thr287Pro mutation in TM2, a domain that contributes to the pore of the GABAA receptors along with corresponding domains of other subunits. Such deficiencies in GABAA receptor functions likely undermine the activity of inhibitory neuronal networks and are consistent with the severe phenotype of EME.
Mutations in different of GABAA receptor subunits, such as α1, β1, β3, γ2 and δ, have been identified in various epilepsy phenotypes.6 ,7 ,15 ,21–33 Mutations in GABRA1 have been identified in early infantile EE and are thought to be associated with childhood absence epilepsy (CAE) and juvenile myoclonic epilepsy.15 ,21 ,24 Mutations in GABRG2 have been found in genetic epilepsy with febrile seizures and are thought to be associated with CAE.27 ,28 Mutations in GABRD have also been identified in GEFS+32 and mutations in GABRB1 and GABRB3 were identified in LGS or IS by the Epi4K consortium.6 Although we reported a mutation in a patient with EME, mutations in GABRB2 may be found in EME or other epilepsy-related phenotypes, as observed in other genes encoding GABAA receptors. Functional studies have examined why mutations in these genes cause various phenotypes. Recently, Janve et al34 published in vitro functional studies of LGS-associated GABRB3 (p.D120N, p.E180G, p.Y302C), IS-associated GABRB3 (p.N110D) and GABRB1 (p.F246S) mutations. The mutations were identified in the Epi4K consortium study. The LGS-associated GABRB3 (p.D120N, p.E180G and p.Y302C) mutations reduced whole-cell currents by decreasing the probability of single channel opening; cell surface receptor expression was normal in these cases. In contrast, the IS-associated GABRB3 (p.N110D) and GABRB1 (p.F246S) mutations caused subtle changes in whole-cell current peak amplitude, but altered current deactivation by decreasing or increasing single channel burst duration, respectively. These molecular and cellular perturbations brought about by these mutations are different from those engendered by the p.Thr287Pro mutant we describe here. These findings suggest that although the diverse phenotypes of patients with epilepsy-associated diseases may depend on the specific GABAA receptor subunit mutation, the heterogeneous clinical consequences of each mutant cannot necessarily be anticipated by in vitro studies.
Although our discovery of a de novo missense mutation of GABRB2 in a child with non-syndromic EME confirms the heterogeneity of EME aetiology, it remains unclear why different GABAA receptor subunit mutations cause a variety of phenotypes, even though they all trigger the same receptor dysfunction (eg, reduction of Cl− current in GABAergic synapses). Perhaps the effect of the mutation is dictated by the precise combination of subunits in the GABAA receptor; this may effectively alter the configuration of GABA receptors in the brain and thus affect the neuronal network. To address these questions, animal models should be used to test the phenotypic effect of novel mutations that are discovered in subunits of the GABAA receptor.
Web resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
ExAC browser, http://exac.broadinstitute.org/
ESP6500, http://evs.gs.washington.edu/EVS/
dbSNP142, http://www.ncbi.nlm.nih.gov/snp/
SIFT, http://sift.jcvi.org/
PolyPhen2 hvar, http://genetics.bwh.harvard.edu/pph2/
Mutation Taster, http://www.mutationtaster.org/
PhyloP100way vertebrate, http://compgen.cshl.edu/phast/
STRING 10, http://string-db.org/
Acknowledgments
The authors would like to thank the patients and their families who participated in this study for their cooperation.
References
Footnotes
Contributors AI and SH conceived and designed the study. Genetic data were generated and analysed by AI. Electrophysiological and protein expression experiments for the mutation were performed by J-QK, CCS, CCH, WS and RLM. JCW tested the distribution of benign variants by statistical method. AI, J-QK, CCS, CCH, WS, JCW, RLM and SH wrote the paper. All authors reviewed the compiled manuscript.
Funding This work was supported by a grant-in-aid for scientific research (A) (24249060 and 151402548)(to SH), grant-in-aid for Challenging Exploratory Research (25670481) (to SH), Bilateral Joint Research Projects (to SH) from Japan Society for the Promotion of Science, grants for Scientific Research on Innovative Areas (221S0002 and 25129708) (to AI and SH) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), MEXT-supported programme for the strategic research foundation at private universities 2013–2017 (to SH), a grant for Practical Research Project for Rare/Intractable Diseases (15ek0109038a) from Japan Agency for Medical Research and development, grant-in-aid for the Research on Measures for Intractable Diseases (H26-Nanji-Ippan-051 and 049) (to SH) from the Ministry of Health, Labour and Welfare, Intramural Research Grant (24-7 and 27-5) for Neurological and Psychiatric Disorders of NCNP (to SH), the Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University (to SH), grants from The Mitsubishi Foundation (to SH) and Takeda Scientific Foundation (to SH), the Kobayashi Magobei Foundation (to AI) and the Kurozumi Medical Foundation (to AI) and the Japan Epilepsy Research Foundation Grant (to AI). The research was also supported by research grants from CURE, Dravet Syndrome Foundation and NINDS R01 062835 (to J-QK) and NINDS R01 NS 33300 (to RLM).
Competing interests None declared.
Patient consent Parental/guardian consent obtained.
Ethics approval Parents of each patient provided signed informed consent using a protocol approved by the Ethics Review Committee of Fukuoka University.
Provenance and peer review Not commissioned; externally peer reviewed.