Article Text

Abstract
Background There are many single-gene causes of steroid-resistant nephrotic syndrome (SRNS) and the list continues to grow rapidly. Prompt comprehensive diagnostic testing is key to realising the clinical benefits of a genetic diagnosis. This report describes a bespoke-designed, targeted next-generation sequencing (NGS) diagnostic gene panel assay to detect variants in 37 genes including the ability to identify copy number variants (CNVs).
Methods This study reports results of 302 patients referred for SRNS diagnostic gene panel analysis. Phenotype and clinical impact data were collected using a standard proforma. Candidate variants detected by NGS were confirmed by Sanger sequencing/Multiplex Ligation-dependent Probe Amplification with subsequent family segregation analysis where possible.
Results Clinical presentation was nephrotic syndrome in 267 patients and suspected Alport syndrome (AS) in 35. NGS panel testing determined a likely genetic cause of disease in 44/220 (20.0%) paediatric and 10/47 (21.3%) adult nephrotic cases, and 17/35 (48.6%) of haematuria/AS patients. Of 71 patients with genetic disease, 32 had novel pathogenic variants without a previous disease association including two with deletions of one or more exons of NPHS1 or NPHS2.
Conclusion Gene panel testing provides a genetic diagnosis in a significant number of patients presenting with SRNS or suspected AS. It should be undertaken at an early stage of the care pathway and include the ability to detect CNVs as an emerging mechanism for genes associated with this condition. Use of clinical genetic testing after diagnosis of SRNS has the potential to stratify patients and assist decision-making regarding management.
- Podocyte
- Steroid-resistant nephrotic syndrome
- Next generation sequencing
- Alport syndrome
- Gene panel testing
- SRNS
- Focal segmental glomerulosclerosis
- FSGS
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
- Podocyte
- Steroid-resistant nephrotic syndrome
- Next generation sequencing
- Alport syndrome
- Gene panel testing
- SRNS
- Focal segmental glomerulosclerosis
- FSGS
Introduction
Patients with nephrotic syndrome (NS) suffer a breakdown of the glomerular filtration barrier at the level of the podocyte leading to massive proteinuria, hypoalbuminaemia and oedema.1 The majority of patients display sensitivity to steroids (steroid-sensitive nephrotic syndrome (SSNS)). Patients with steroid-resistant nephrotic syndrome (SRNS) account for 5%–10% of cases.2 Patients with SRNS often need invasive renal biopsy to determine characteristic histological features of the disease such as focal segmental glomerulosclerosis (FSGS), and many progress to end-stage renal disease within 5 years.3 4
Pathogenic variants in single genes affecting podocyte function are a common cause of SRNS, reported in up to 29.5% of a predominantly childhood cohort.5 There is a differing spectrum of disease genes and pathogenic variants associated with congenital and childhood-onset disease in comparison with adult disease, and genes may present a renal-only phenotype or NS as part of a wider syndrome. Alport syndrome (AS) is associated with pathogenic variants in COL4A3, COL4A4 or COL4A5 and may present with proteinuria (some with FSGS on biopsy), and more commonly haematuria.6 The renal histology is characterised by an alteration of the glomerular basement membrane. Variants in COL4A3 and COL4A4 have been identified in association with FSGS, together with thin basement membrane nephropathy but without the extrarenal features of AS.7
The proportion of single-gene cases identified inversely correlates with the age of onset with 69.4%–100% of congenital-onset disease reported as having a genetic aetiology.5 8 Over 53 genes, both recessive and dominant, have been associated with SRNS.9 10 The phenotypic spectrum is widening, both in terms of age of onset11 and phenotypic variability, with novel phenotypes for individual genes emerging.12 Recent evidence suggests that the phenotype of a pathogenic variant in an SRNS gene can be modified by a variant in one of the collagen genes (COL4A3, COL4A4, COL4A5).13 14 The phenotype may also depend on the location of the variant within the gene/protein,15 and whether single or multiple variants are present in one or more genes.16
Timely genetic testing can considerably alter patient management and facilitate a greater understanding of the genetic complexity of the condition.17 Recent studies report a multiple-gene testing approach using next-generation sequencing (NGS).5 13 18 19 Only a single NGS study has reported copy number variants (CNV) in NS genes,10 hence the contribution of this mechanism to SRNS is currently largely unknown, although evidence has suggested this as a mechanism.20 21 Several studies report patients with a typical phenotype and only a single recessive pathogenic variant.22 23 This suggests there may be as yet uncharacterised variants and that a comprehensive NGS assay with the ability to detect CNVs would be of increased value.
We have developed a clinically approved gene panel test for 37 SRNS and collagen-related genes (table 1) using a targeted amplicon-based NGS assay and bespoke bioinformatics analysis that detects both single-nucleotide variants (SNVs) and CNVs in batches of 12–16 patients. Importantly, the panel has the flexibility to extend according to discovery of new genes. An enlarged panel of 70 renal-associated genes has been offered since March 2017 for new referrals including novel genes recently reported associated with SRNS such as NUP93, NUP107, NUP205, KANK1, KANK4, MAGI2, EMP2 and ANLN.24–28
Genes included in the diagnostic 37-gene panel and coverage
Methods
In total, 302 patients were referred with informed consent for diagnostic gene panel analysis over a 26-month period. The diagnostic test has been formally assessed for validity, and socio-legal/ethical implications by the UK Genetic Testing Network and UK National Health Service (NHS) commissioners through the gene dossier process, and was undertaken in an accredited UK NHS Laboratory. Data presented pertain only to anonymised auditing of routine diagnostic testing; therefore, this study was not subject to ethical approval.
The sequencing and bioinformatics protocol is described in brief with further details as supplementary material. A custom HaloPlex Target Enrichment System (Agilent) was designed to target 37 genes associated with SRNS (table 1). Samples were sequenced on a MiSeq (Illumina) analyser following the manufacturer’s protocol. Bioinformatic analysis was performed using a bespoke pipeline based on the Broad Institutes’ Best Practice guidelines.29 Variants were classified according to the Association for Clinical Genetic Science (ACGS) best practice guidelines (see online supplementary table 1).30 In subsequent discussion, Class 4 and Class 5 variants are grouped together and termed ‘likely-pathogenic’ (LP). Class 3 are variants of unknown clinical significance (VUS). Class 3–5 variants were confirmed by Sanger sequencing.
Supplementary file 1
Supplementary file 2
Where possible, relative testing was undertaken using Sanger sequencing to determine phase (cis or trans) and segregation. CNVs in patients with single heterozygous variants in recessive genes were identified by CONTRA31 and confirmed using multiplex ligation-dependent probe amplification (MLPA).
Clinical data were supplied by a proforma. This included a question to gauge potential management changes as a result of the genetic analysis: ‘Will the result from this genetic test lead to a change in immunosuppression?’ Response to this was at individual clinician’s discretion and was not mandatory for processing of samples.
Results
Demographics
The majority of patients had a clinical diagnosis of idiopathic nephrotic syndrome, mostly SRNS, but 12 were SSNS either frequently-relapsing or steroid-dependent. Thirty-five patients were referred with features suggestive of AS including haematuria, a family history, hearing loss or thin basement membrane on biopsy. For clinical analyses, we have therefore separated the cohort into SRNS, SSNS and Alport groups. Referrals were received from 12 different countries (see online supplementary figure 1). The timing of disease onset was known for 196 patients: 32 (16.3 %) were congenital (<3 months), 16 (8.2%) infantile (3–12 months), 101 (51.5%) childhood (1–12 years), 17 (8.7%) juvenile (13–18 years) and 30 (15.3%) adult (>18 years). Of 255 patients with SRNS, a biopsy report was available in 133 which showed FSGS in 109 (82.0%) and minimal change disease (MCD) in 8 (6.0%). In 9 of 12 patients with SSNS with a biopsy report, 3 (33.3%) had FSGS and 3 (33.3%) had MCD. In patients with SRNS, 35 (23.8%) of 147 with data available had a family history of renal disease. Among 132 patients with SRNS where data on age of onset and family history were available, 52.6% (10/19) of adults compared with 12.4% (14/113) of patients<18 years had a positive family history. This may represent differing referral patterns in clinicians caring for adult patients with NS such that they were less likely to request genetic testing for adults without a family history. Among 35 patients in the Alport group, 78.6% of 28 patients with data had a family history of a similar disease. Other demographic data are shown in table 2.
Supplementary file 3
Clinical characteristics of cohort
Data quality and gene panel performance
An average gene coverage of 99.26% coding sequence at a minimum read depth of 30× was achieved on a typical 12-patient run. The per-gene coverage is shown in table 1. The ACGS reporting time guideline for large panel tests is 112 calendar days.32 The median time from receipt of samples to issue of a report was 74 days (IQR 49–106 days). With clinically urgent referrals, it was possible to reduce substantially the turnaround time with complete genetic panel reports provided for 17 patients within 4 weeks and the fastest positive case report (NPHS2 compound heterozygote) being issued in 22 calendar days.
Genetic variants
Targeted gene panel testing of all 302 patients identified 71 (23.5%) with a likely genetic cause for disease. The genetic diagnostic rate among the group with SRNS was 21.2%, including 44/209 (21.1%) paediatric and 10/46 (21.7%) adult nephrotic cases (figure 1). In patients with SRNS where family history was known, the genetic diagnostic rate was 11/35 (31.4%) in those with a positive family history and 30/112 (26.8%) in those with negative family history. In patients with SRNS where family history was known and with age of onset over 18 years, the genetic diagnostic rate was 2/10 (20.0%) in those with a positive family history and 3/9 (33.3%) in those with negative family history. The rate in those with parental consanguinity was 5/13 (38.5%) compared with 28/104 (26.9%) in those without. In all the 12 patients with SSNS, no pathogenic variants were found in any of the 37 genes tested. The genetic diagnostic rate was 48.6% for the Alport group.

Flow chart of patients by phenotype at presentation, age and genetic diagnosis. ‘Age’ refers to age at diagnosis or, if not available, age at genetic testing. ‘Genetic’ refers to the number of cases with likely pathogenic variants. SRNS, steroid-resistant nephrotic syndrome; SSNS, steroid-sensitive nephrotic syndrome.
The spectrum of pathogenic variants is summarised in table 3. Detailed phenotypic and variant data for the 71 patients with genetic disease is shown in online supplementary table 2.
Genes with likely pathogenic variants in the steroid-resistant nephrotic syndrome group and haematuria/Alport syndrome group
The most frequently detected LP variants in the SRNS group (n=255) were in NPHS1, WT1 and NPHS2 in 12 (4.7%), 11 (4.3%) and 7 (2.7%) patients, respectively. In the Alport group, the variants were all in collagen genes: COL4A3, COL4A4 and COL4A5 in 5 (14.3%), 2 (5.7%) and 10 (28.6%) patients, respectively. Of note, five SRNS/FSGS patients had LP variants in COL4A3, COL4A4 and COL4A5 (one, one and three patients, respectively) including a single novel LP COL4A3 variant, c.698G>A, p.(Gly233Glu), in a patient with a dominant family history of FSGS (patient 4) which tracked with disease in an affected brother. Among patients with SRNS/FSGS who were found to have genetic disease (excluding those with collagen variants), there was an autosomal-dominant mode of inheritance in 16/41 (36.6%) of those with disease onset ≤18 years compared with 6/7 (85.7%) of those >18 years.
Variants of unknown significance
In addition to the patients with LP variants, a further 40 patients had one or more VUS (see online supplementary table 3). Among the 52 VUS in these patients, the most frequently involved genes were: 28.9% in collagen genes; 7.7% each in NPHS1 and NPHS2; and 5.8% each in INF2, MYH9, PLCE1 and PTPRO. Of the 71 patients with likely genetic disease, 11 cases had 12 additional VUS in genes other than the main causative one for that patient, most frequently collagen genes in 41.7% and WT1 in 16.7%. Overall, of the 64 recorded VUS, 31.3% were in collagen genes, 6.3% in NPHS2, 6.3% in NPHS1 and 6.3% in MYH9.
Novel variants
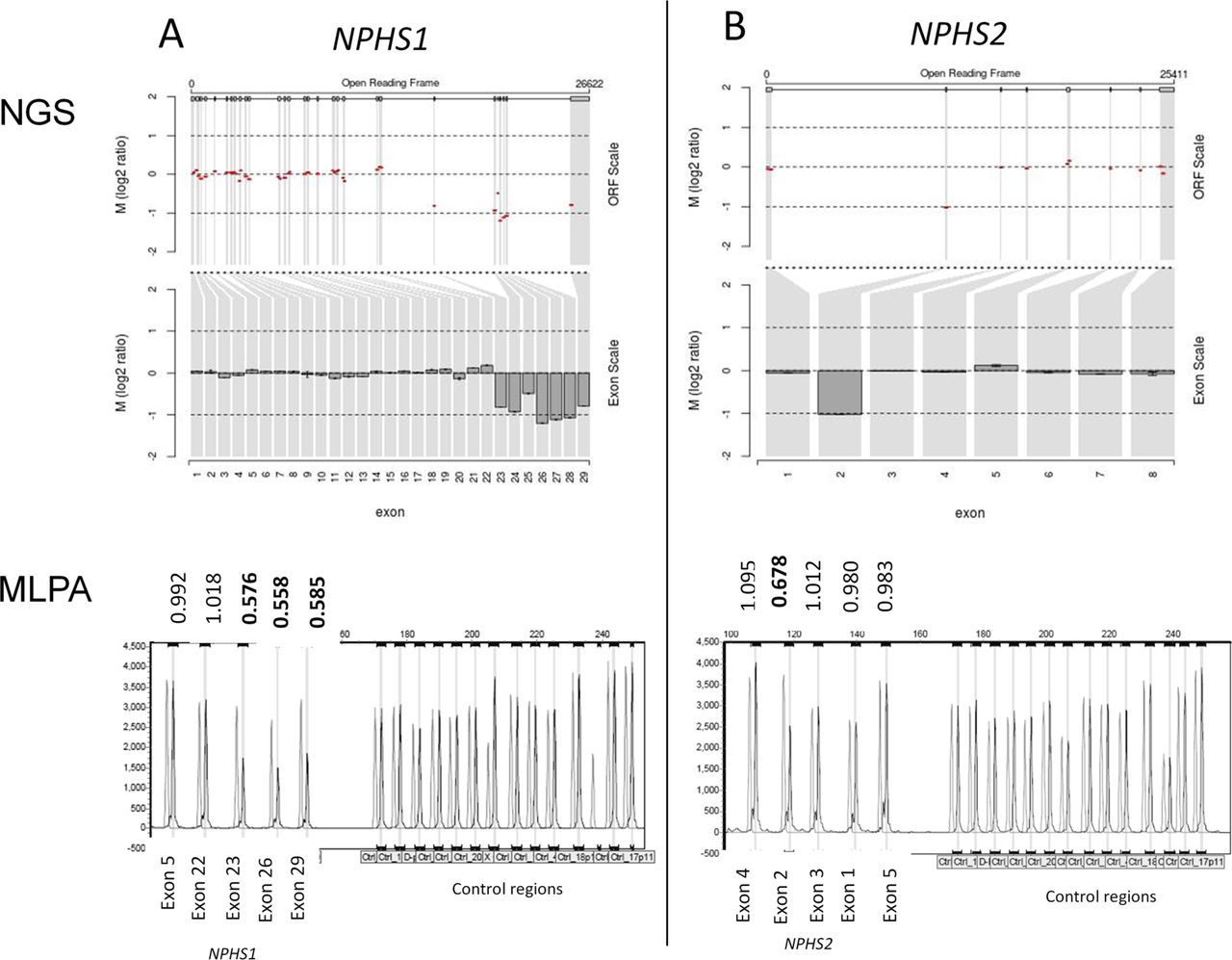
Among the 71 patients with a genetic cause for disease, 32 had variants without a previous disease association including 26 with one or more novel variants absent from population databases. Two patients had gene deletions of one or more exons detected by CNV analysis (figure 2). Patient 44 (see online supplementary table 2) presented with congenital nephrotic syndrome (CNS) and had a maternally inherited truncating deletion of NPHS1 exons 23–29 together with a paternally inherited previously-reported nonsense variant c.866G>A p.(Trp289*).33 Patient 55 also presented with CNS and genetic testing revealed a maternally inherited frame shift deletion of NPHS2 exon 2 in combination with a paternally inherited c.1032delT variant. The c.1032delT variant has been previously reported as the most frequent pathogenic variant in NPHS2 in Poland (Kashubian region).23 Both parents of patient 55 are of Polish extraction. In addition to these two patients, a further 30 had variants without a previous disease association in the following genes: ACTN4 (1 patient), COL4A3 (2), COL4A4 (1), COL4A5 (10), INF2 (2), LAMB2 (2), NPHS1 (5), NPHS2 (2), SMARCAL1 (1), TRPC6 (1) and WT1 (3).
![[SP3.jpg]](https://jmg.bmj.com/content/jmedgenet/54/12/795/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
{kind=link}
NPHS1 and NPHS2 copy number variants. (A) NPHS1 deletion of exons 23–29. (B) NPHS2 deletion of exon 2. Next-generation sequencing (NGS) read depth analysis (top) with results confirmed by MLPA (below). NGS fold change in copy number (Log2 ratio) is shown across the locus (Open Reading Frame (ORF) scale) and averaged for each exon (exon scale). MLPA shows patient peaks in dark grey and normal control peaks in light grey. The patient/normal ratios are shown with deletions having ratio <0.75.
Novel INF2 LP variants were detected in two adult-onset NS patients: p.(Tyr50Asp) (patient 25) and p.(Leu165Arg) (patient 26). Both were missense variants affecting highly conserved residues within the diaphanous inhibitory domain of the INF2 protein consistent with the previously reported spectrum of disease-causing variants. Segregation supports pathogenicity, p.(Tyr50Asp) co-segregating with disease in five affected family members and p.(Leu165Arg) present in one affected family member and absent in two unaffected family members. An additional sensory neuropathy phenotype previously reported in 12.5% of INF2 cases34 was also seen in affected family members with the p.(Tyr50Asp) variant.
Two novel missense WT1 variants were identified in the known hotspot region (exons 6–9)35 in patients with atypical presentation and no recorded extrarenal manifestations. p.(His339Arg) in exon 7 (patient 61) co-segregates with disease in six affected family members with a variable phenotype ranging from childhood-onset nephrotic-range proteinuria to mild proteinuria presenting in adulthood. The p.(Arg390Gln) variant in WT1 exon 8 (patient 65) was associated with age of onset of 30 years and was inherited from an affected father who was diagnosed in his 30s. Although WT1 is normally associated with childhood-onset disease, these findings are consistent with a previously described biphasic childhood and adulthood presentation of variants in WT1.5
Single heterozygous variants in recessive NPHS1 and NPHS2
In six clinically affected SRNS cases (patients 112–117 in online supplementary table 4), full coding sequence analysis detected a single heterozygous pathogenic variant in NPHS1 or NPHS2 that has been previously published as disease causing. No CNVs were identified. The finding of these variants may be incidental. However, given the low incidence of SRNS, the young age of onset (<5 years, two with CNS) and the low frequency/absence of the variants in databases of subjects without known renal disease it is possible that there are additional NPHS1/NPHS2 variants in unsequenced intronic or promoter regions which may act in combination to cause the phenotype in these patients. These findings were reported as compatible with the phenotype but insufficient to make a diagnosis.
Three of these patients (112, 113 and 114) with early-onset SRNS were heterozygous for previously reported rare pathogenic variants in NPHS1. Patient 112 developed SRNS under the age of 4 years with a heterozygous p.(Asp105Asn) variant previously reported in a Japanese patient with CNS where a second variant was not detected.36Patient 113 had a heterozygous p.(Arg299Cys) variant. Patient 114, from a Jordanian consanguineous family, who presented with CNS and FSGS on biopsy had a c.1138C>T, p.(Gln380*) nonsense variant but, in common with the other cases, no other likely pathogenic variants in NPHS1 or other genes in the panel. Patient 117 had a single heterozygous missense variant in NPHS2: c.872G>A, p.(Arg291Gln) previously reported as pathogenic in the homozygous/compound heterozygous state. Patient 117 also had a novel single heterozygous variant in NPHS1: c.2512C>A, p.(Pro838Thr) which is not reported in population databases and prediction tools suggest is deleterious.
Two further patients (115 and 116) presenting with classical NS had single previously reported pathogenic variants in NPHS2 p.(Arg138Gln) (exon 3) and p.(Leu156Phefs*11) (exon 4) in a compound heterozygous state with the NPHS2 non-neutral polymorphism p.(Arg229Gln). Tory et al 15 previously demonstrated that p.(Arg229Gln) is only pathogenic in combination with variants in exons 7 or 8 and therefore should not be pathogenic with p.(Arg138Gln) or p.(Leu156Phefs*11).15 It is possible that a third intronic or promoter variant in NPHS2 is contributing to these patients’ phenotypes.
Likely pathogenic variants by age of disease onset
The age of disease onset was known with certainty for 164 (64.3%) of 255 patients referred with a clinical presentation of SRNS and online supplementary figure 2 illustrates the genetic diagnostic rates by age group. In patients with CNS, the diagnostic rate was 58.1% (18/31) with LP variants in the following genes: NPHS1 (12 patients), LAMB2 (3 patients), NPHS2 (2 patients) and WT1 (1 patient). For cases of SRNS with known age of onset >18 years, the rate was 28.6% (6/21) with LP variants in INF2 (two patients), WT1 (two patients), SCARB2 (one patient) and TRPC6 (one patient).
Supplementary file 4
![[SP4.jpg]](https://jmg.bmj.com/content/jmedgenet/54/12/795/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
Clinical impact of genetic testing
Physicians were asked whether the results of genetic testing would alter immunosuppressive management in patients with SRNS. Responses were obtained in relation to 71 (27.8%) of these patients. The response rate was not high enough to make definitive conclusions, but broadly clinicians reported that results would influence decisions to reduce or stop immunosuppression and one physician indicated that treatment decisions would be made after the gene panel results were known. The diagnostic test result from 67 patients resulted in the subsequent testing of 148 family members including two prenatal tests for LAMB2 and NPHS2 pathogenic variants. At least nine family tests are known to have helped inform suitability for donor transplant treatment. Other familial testing has provided diagnoses for relatives and aided in clarifying variant pathogenicity. Other responses confirmed that genetic testing impacts on diagnosis and prognosis after transplant.
Discussion
Gene panel testing is becoming more and more relevant for screening rare diseases, with greatly improved cost/benefit. The knowledge of the genetic basis of SRNS has expanded considerably over the past five years with several national and international cohorts recently published5 10 37 where genetic analysis was undertaken either as part of a research study or on a limited number of genes dependent on the country or institution where the patient was seen. This study reports on large gene panel testing available on a clinical diagnostic basis within the NHS in the UK. Analysis is performed at a single accredited centre, with technological and bioinformatics expertise developed over several years in collaboration with academic research institutes. Although the majority of referrals are received from UK clinicians, 41% of referrals are from outside the UK.
The frequency of likely pathogenic variants among patients with SRNS was 21.2%. This is marginally lower than 24%–34% reported in other studies which included predominantly subjects with childhood-onset disease.5 8 19 37 The cohort reported here includes patients referred for genetic testing by clinicians in routine practice and is therefore more heterogeneous than those included in international registries of SRNS. The cohort was also not restricted to patients with childhood-onset disease with 30 patients having known onset in adulthood. As illustrated in online supplementary figure 2, the genetic diagnostic rate decreased with increasing age of onset from congenital to childhood, similar to that reported in another international cohort.5 However, there was an increased diagnostic rate in the juvenile and adult subgroups. It is likely that the adults referred by clinicians for gene panel testing are a filtered population of cases as suggested by the higher frequency of a positive family history of 52.6%.
Twelve patients with SSNS were referred for genetic testing and none had a potentially pathogenic variant. Thus far, no purely monogenic causes for SSNS have been identified.38 Associations between SSNS and variants in EMP2, KANK1 and KANK2 have been described in family studies but evaluation in larger cohorts did not identify additional patients with pathogenic variants.25 27 KANK2 was included in the gene panel reported here and no likely pathogenic variants were identified. The updated version of the panel includes EMP2, KANK1 and KANK4. Our current recommendation for clinicians is not to use the NGS gene panel test for patients with persistently SSNS unless there are specific reasons to suspect a genetic aetiology.
In this study, 100% of LP variants identified by NGS meeting diagnostic variant-calling quality parameters have subsequently been confirmed as being present on Sanger sequencing. Sanger confirmation of NGS LP variants is currently in line with best practice ACGS guidance. It also provides a confirmation of sample identity following pooling of samples during the NGS process and establishes a familial test for relatives. As further evidence is collected, confirmatory testing may become redundant.
The use of gene panel testing supersedes stepwise screening protocols8 and avoids phenotype selection bias, allowing detection of pathogenic variants in genes that would not necessarily be expected from the clinical presentation such as the two adult cases with a WT1 variant without any manifesting extrarenal features. In addition, variants in secondary genes which may potentially contribute to the phenotype of the patient can be identified by a panel approach.
Gene panel screening identified 32 likely pathogenic variants without a previous disease association. Absence or rarity in population data is used as evidence to support pathogenicity; however, it is acknowledged that some ethnicities in this global cohort were either not known or may currently be insufficiently represented in population databases. Where possible, segregation analysis was performed to provide additional evidence to support pathogenicity; however, family members were not always available for testing, reflecting the use of this panel in a clinical setting.
The interpretation of variants of unknown significance in a global cohort is also a challenge of panel testing. As well as segregation analysis, future expansion of population databases will allow improved filtering of population-specific variants and functional work may also aid the interpretation of variants.
This study has demonstrated two cases of CNVs present in NS genes; therefore, CNV analysis of NGS data should be routinely undertaken as part of the variant analysis pipeline, together with confirmation using a second method such as MLPA. It is also apparent that there are a number of clinically typical cases with only a single known NPHS1 or NPHS2 pathogenic variant detected, suggesting deep intronic or regulatory variants if these genes are truly recessive in mechanism. Future whole-genome sequencing in these patients may help to elucidate a genetic pathogenesis.
The timing of testing in relation to disease onset and the speed of genetic reporting are important for clinical utility. It is potentially possible to generate results within 1–2 weeks, thereby avoiding diagnostic biopsy in some cases. In certain contexts, earlier testing and more rapid turnaround are important because results may have direct consequences for prenatal testing and patient treatment. Pathogenic variants in COQ2, COQ6 or PDSS2, coding for proteins the coenzyme Q10 pathway, may indicate the potential for benefit from treatment with this enzyme.39 40 Identification of a causative variant may lead to clinicians stopping or avoiding intensification of immunosuppressive treatment. There has, however, been a report of a patient with NS, diffuse mesangial sclerosis and PLCE1 variants who responded to treatment with steroids and ciclosporin.41 Cases of unaffected older children and adults with the same homozygous PLCE1 variants as their affected relatives suggest a more complex genotype–phenotype interplay and raise the possibility of spontaneous improvement rather than a true response to medication.42 43 Some patients with WT1 variants have responded to steroids and immunosuppression.44 Certain pathogenic variants, such as in WT1, should prompt search for other features of an associated syndrome, such as Frasier syndrome and risk of gonadoblastoma.45
The presence of a causative variant in SRNS is associated with a lower risk of post-transplantation recurrence of disease, occurring in 25.8% of patients testing negative for genetic disease compared with 4.5% of those with an identified variant in a European cohort37 and 0% in a published UK cohort.10 Availability of results supporting a genetic diagnosis may prompt more rapid progression to potentially definitive treatment with transplantation rather than persisting with partially effective medical therapies. Targeted sequencing of family members resulting from gene panel testing has been used prior to transplantation, particularly in cases with autosomal-dominant gene variants.
We report that NGS gene panel testing with bioinformatics analysis for SNVs and CNVs at an early stage after diagnosis of SRNS or suspected AS with results in a clinically relevant time scale has the potential to improve patient stratification and the care pathway.
References
Footnotes
Contributors MW and MAS conceived and designed the study. ESS, PD and LY-S collected and analysed the clinical data. PD, LY-S, AB, GW, CB, GD and MW performed the genetic analysis. ESS and PD created the tables and figures. ESS, PD and MW wrote the first draft of the manuscript. ESS, PD, AB, GIW, MW and MAS were involved in manuscript editing. All authors read and approved the final version.
Funding ESS is funded by a National Institute for Health Research (NIHR) Rare Diseases Translational Research Collaboration (RD-TRC) Clinical Research Fellowship. AB was supported by Kids Kidney Research and Kidney Research UK.
Disclaimer The views expressed are those of the authors and not necessarily those of the UK National Health Service, the NIHR or the Department of Health.
Competing interests MAS has external consultancy roles with UCB, Retrophin and Pfizer. All other authors declare no conflicts ofinterest.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.