Article Text

Abstract
The mammalian or mechanistic target of rapamycin (mTOR) signalling pathway has multiple roles in regulating physiology of the whole body and, particularly, the brain. Deregulation of mTOR signalling has been associated to various neurological conditions, including epilepsy. Mutations in genes encoding components of Gap Activity TOward Rags 1 (GATOR1) (DEPDC5, NPRL2 and NPRL3), a complex involved in the inhibition of the mTOR complex 1 (mTORC1), have been recently implicated in the pathogenesis of a wide spectrum of focal epilepsies (FEs), both lesional and non-lesional. The involvement of DEPDC5, NPRL2 and NRPL3 in about 10% of FEs is in contrast to the concept that specific seizure semiology points to the main involvement of a distinct brain area. The hypothesised pathogenic mechanism underlying epilepsy is the loss of the inhibitory function of GATOR1 towards mTORC1. The identification of the correct therapeutic strategy in patients with FE is challenging, especially in those with refractory epilepsy and/or malformations of cortical development (MCDs). In such cases, surgical excision of the epileptogenic zone is a curative option, although the long-term outcome is still undefined. The GATOR1/mTOR signalling represents a promising therapeutic target in FEs due to mutations in mTOR pathway genes, as in tuberous sclerosis complex, another MCD-associated epilepsy caused by mTOR signalling hyperactivation.
- Focal Epilepsy
- DEPDC5
- GATOR1
- mTOR
- Malformations of Cortical Development
Statistics from Altmetric.com
Introduction
Mammalian or mechanistic target of rapamycin (mTOR) is an ubiquitously expressed serine/threonine kinase regulating cell growth, proliferation, metabolism, motility, death (through apoptosis and autophagy), protein synthesis and transcription.1–3 Brain-specific roles include regulation of synaptic plasticity and learning, neurogenesis and dendritic/axonal morphology.4–7 The protein is a catalytic component of two different complexes, mTORC1 (mTOR complex 1) and mTORC2 (mTOR complex 2), in which mTOR associates with two regulatory subunits: Raptor and Rictor, respectively (figure 1).3 mTORC1 represents one of the most important regulators of cell growth.1 The activity of this complex is controlled by numerous factors (insulin, growth factors, amino acids and oxidative stress)8 ,9 acting through various protein signalling pathways (figure 1).6 One of these pathways recruits the GATOR (Gap Activity TOward Rags) complex, involved in the amino acid sensing activity (figure 1).10 GATOR complex is composed of two subcomplexes, GATOR1 and GATOR2, which function as negative and positive regulators of mTORC1, respectively (figure 1).10–13 The former includes DEPDC5 (DEP domain-containing protein 5), NPRL2 (nitrogen permease regulator 2-like protein) and NPRL3 (nitrogen permease regulator 3-like protein) proteins, the latter MIOS (missing oocyte, meiosis regulator, homologue), SEH1L (SEH1 like), SEC13 (SEC13 homologue), WDR24 (WD repeat domain 24) and WDR59 (WD repeat domain 59). The role of each protein in these complexes has still to be fully clarified, but the conservation of the mTOR pathway architecture across different species suggests that each partner of the complex carries out fundamental functions.14 Recently, mutations in the genes encoding the components of the GATOR1 subcomplex have been associated with several genetic focal epilepsy (FE) syndromes, including autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE, MIM 600513), recently renamed as autosomal-dominant sleep-related hypermotor epilepsy;15 epilepsy with auditory features (EAF, also known as lateral temporal lobe epilepsy, TLE, MIM 600512); familial FE with variable foci (FFEVF, MIM 604364).16–20 DEPDC5 mutations have also been recently described in patients manifesting rolandic epilepsy or epileptic spasms.21 ,22

Schematic representation of mTOR signalling pathway. mTOR, mammalian or mechanistic target of rapamycin.
Mutations of GATOR1 in FE syndromes
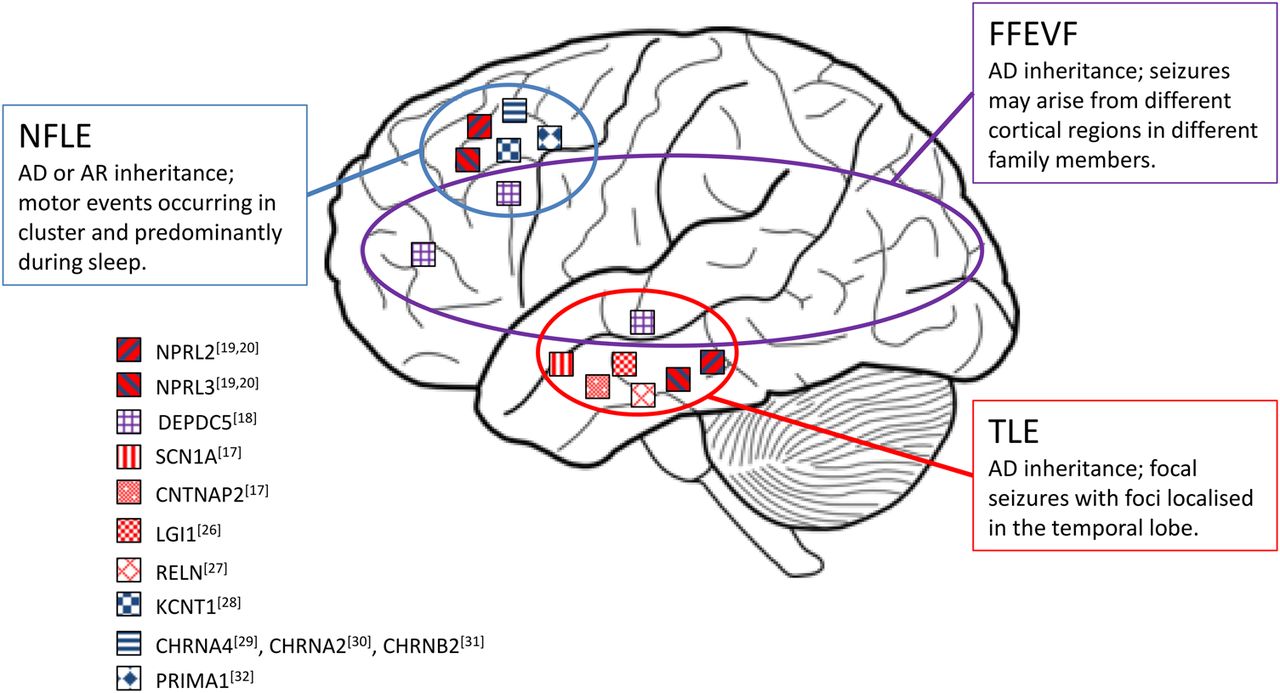
Epilepsy is a widespread neurological disorder affecting about 1% of the global population.23 In FE, seizures originate from a limited area of the brain (the epileptogenic zone, EZ). FEs account for nearly 60% of all epileptic syndromes, and can be caused by genetic as well as acquired factors.24 ,25 Paradigmatic FE genetic syndromes (EAF, ADNFLE and FFEVF) have been associated with mutations in a number of genes (figure 2) coding for proteins implicated in different brain functions (eg, synapse development and transmission or neuronal excitability).17–20 ,26–32 The identification of mutations in GATOR1 genes in all these epileptic syndromes opens the interesting scenario of defects of the same pathway underlying different epilepsy phenotypes (figure 3 and table 1 provide a summary of the mutations identified so far).16–18 ,20 ,21 ,33–41
Published GATOR1 gene mutations and associated phenotypes.
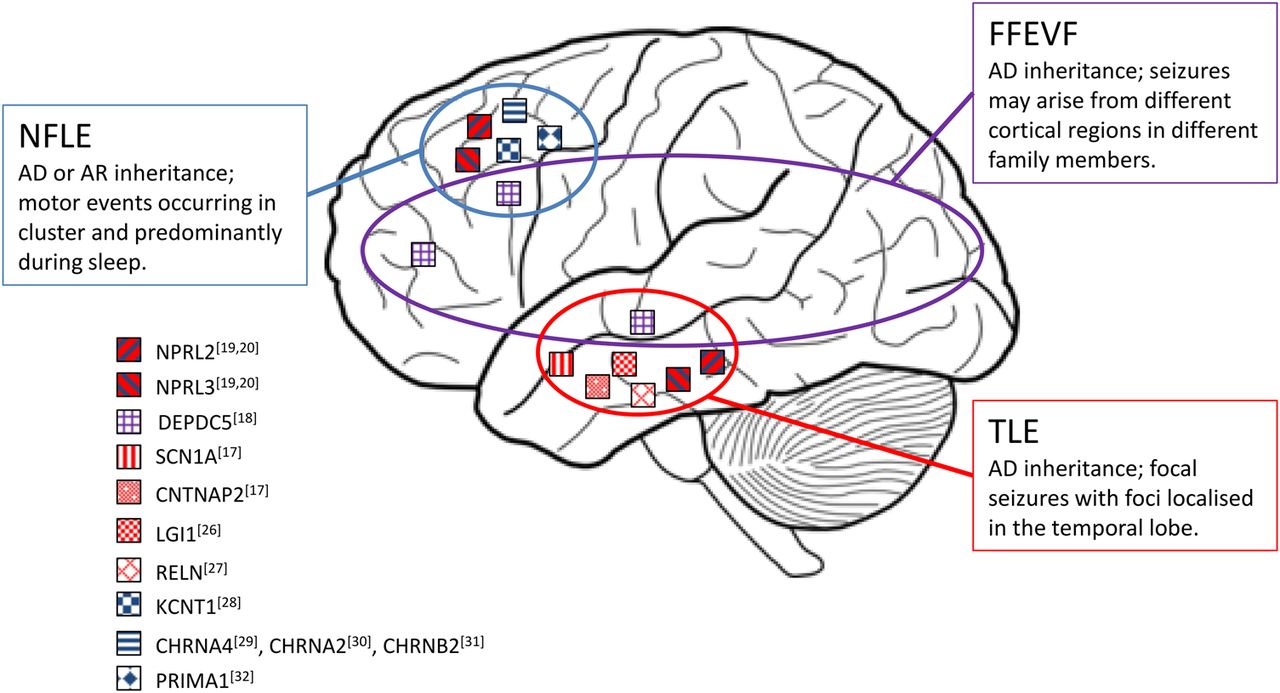
Genes mutated in the major focal epilepsy syndromes. The localisation of the squares on the brain image is related to the cortical lobe involved in the epileptic discharges in the major FEs to which these genes have been associated. AD, autosomal dominant; AR, autosomal recessive; NFLE, nocturnal frontal lobe epilepsy; TLE, temporal lobe epilepsy; FFEVF, familial focal epilepsy with variable foci.

{kind=link}
{kind=link}
{kind=link}
Schematic representation of Gap Activity TOward Rags 1 (GATOR1) genes and published mutations. The mutations reported are subdivided in loss-of-function mutations (upper panel in each gene representation) and missense mutations (lower panel). Splicing affecting mutations listed in table 1 are not included.
Mutations in DEPDC5, first identified in seven out of the eight FFEVF families linked to chromosome 22q12,18 were subsequently implicated in the 5%–37% of a broad range of FEs, including NFLE and TLE.16 ,36 More recently, the description of NPRL2 and NPRL3 mutations in patients with FE confirmed the involvement of the entire GATOR1 complex in the pathogenesis of different FE syndromes.19 ,20 Collectively, the data published so far indicate that mutations in genes encoding the GATOR1 complex are the most frequent genetic cause of FEs, globally accounting for 9% of the cases.20 Mutations in these genes may show a wide interfamilial and intrafamilial variability in epilepsy phenotypes, as exemplified by FFEVF. Within a family, the same mutation can be associated with lesional and non-lesional epilepsy: in only some of the affected members, brain MRI disclosed malformations of cortical development (MCDs), in particular focal cortical dysplasia (FCD) type IIb.19 ,20 ,38–41 FCDs are the most common cause of medically refractory epilepsy.45 Surgery represents a highly effective treatment in these patients and requires a comprehensive presurgical assessment for identification of the EZ, especially in the 15% of ‘lesion negative’ cases where subtle FCD are detected only by histology.46 The aetiology of FCDs is largely unknown, but the identification of mutations in GATOR1-encoding genes in patients with such MCDs suggests an alteration of mTOR signalling pathway. Interestingly, other forms of MCDs are due to abnormal neuronal and glial proliferation caused by an aberrant activation of mTOR signalling cascade, as in tuberous sclerosis complex (TSC), which is caused by mutations in TSC1 and TSC2, encoding two interacting inhibitors of mTORC1 (figure 1).47 Furthermore, recent publications have demonstrated that somatic brain mutations are implicated in the pathogenesis of MCDs, as already recognised for tumour development in patients with TSC.40 ,41 ,48–51 In FE families, MCDs may be observed in only a portion of the affected members carrying a germinal mutation in a GATOR1-encoding gene.38 ,40 ,41 This might be explained by the limitations of conventional neuroimaging techniques, which may fail to detect subtle brain lesions.46 Alternatively, a ‘two-hits model’ has been proposed, by which a second brain somatic mutation in the same gene is hypothesised to be responsible for the development of MCD. Baulac et al40 studied a family, in which a DEPDC5 mutation segregated in all affected members. In one of the patients with FCD, they examined a lesional tissue specimen and found a brain somatic DEPDC5 mutation in addition to the germline hit. They suggested that this is consistent with a two-hit model leading to biallelic inactivation of the gene within the lesional tissue. However, there is still limited experimental evidence that this hypothesis, which has been proven valid for TSC1/TSC2,51 holds for genes of the GATOR1 complex. Most of the DEPDC5 mutations described so far are loss-of-function mutations (table 1), suggesting that the loss of the inhibition of mTORC1 activity could be the cause leading to epilepsy. The hyperactivation of mTORC1 has been also detected both with in vitro systems and in brain resections from GATOR1 mutated patients.19 ,38 ,42 This latter point is of particular interest because an abnormal mTORC1 signalling is known to cause alterations in neuronal migration and cortical lamination, as reported in tuberous sclerosis52 and as documented in the heterozygous DEPDC5 rat model (Depdc5±) recently published.53 The functional assessment of 12 selected DEPDC5 variants identified in patients with FE has revealed that only a portion has a clear effect on DEPDC5 signalling and mTORC1 activation, in terms of expression, GATOR1 complex formation, and interaction with active RagA/B-RagC/D heterodimers in vitro (see below). These findings then suggest that some of the identified GATOR1 variants could have distinct consequences on GATOR1 function that may explain the phenotypic variation observed among patients.42 Consequently, the finding of a hyperactivation of mTORC1 signalling in patients with GATOR1 mutations suggests a novel common pathological mechanism underlying FEs with or without MCDs: indeed these could be included in the so-called ‘mTORopathies’, with important implications both in patients' treatment and prognosis. However, since MCDs are not frequently found in GATOR1-associated epilepsies, further studies will be needed to elucidate the mechanism underlying MCD formation.
GATOR1 subcomplex features and its role in the mTOR pathway
DEPDC5 is located on chromosome 22q12 and encodes a 1603–amino acid protein, ubiquitously and constantly expressed both in the developing and in the adult brain, and characterised by two functional domains.18 The DUF3608 domain of the protein is thought to contribute to the interaction of the DEPDC5 homologue with the two other components of GATOR1 in yeast,54 but its function has not been characterised in mammals yet. The DEP domain, which was named after the initials of the proteins Disheveled, Egl-10 and Pleckstrin, is a globular domain found in numerous GTPase activating proteins. The DEP domain is also found in DEPTOR (a subunit of mTORC1) where it has a role in the interaction between RGS (regulator of G protein signalling) proteins and their membrane-bound G-protein-coupled receptors.14 ,55 NPRL2 and NPRL3 are located on chromosomes 3p21.3 and 16p13.3 and encode two proteins of 380 and 569 amino acids, respectively. Both proteins are characterised by N-terminal longin domains and PEST motifs.14 Longin domains are usually found in guanine nucleotide exchange factor (GEF) proteins; however, a GEF activity has not yet been demonstrated for these two proteins.56–58 On the other hand, PEST motifs are often found in rapidly degraded proteins, although these are not well conserved in mammals and could not elicit this function in NPRL2/3.11 ,14 No three-dimensional (3D) map of the GATOR1 complex is available at present, but a recently published combination of biochemical and computational approaches has revealed the first 3D map of the yeast homologue of the mammalian GATOR complex, in which the two homologous subcomplexes form connected discrete modules, suggesting similar interactions between GATOR1 and GATOR2.14 ,59 The GATOR complex is a key regulator of the cellular sensing of nutrients levels, through its regulation of mTORC1 activity, which plays a pivotal role in this signalling; however, this has not been well studied in neural systems.1 ,60 Figure 1 illustrates the main signalling cascades converging on mTOR regulation. In the past few years, GATOR1 was implicated in the inhibition of Rag GTPase heterodimers RagA/B-RagC/D, which are involved in mTORC1 recruitment at the lysosome membrane, a key step required for its phosphorylation and consequently activation by the small GTPase RHEB.10 ,61 GATOR1 inhibition of RagA/B-RagC/D is blocked by GATOR2 activation.10 The outcome of the fine regulation of mTOR activity in the brain is the control of brain development and function.4–7
Neurological disorders related to mTOR pathway deregulation
Given the multiple roles of mTOR pathway in brain homeostasis and development, it is not surprising that its deregulation has been implicated in several monogenic neurodegenerative and neuropsychiatric diseases. These include the already cited TSC, caused by mutations in TSC1 and TSC2, or the PTEN Hamartoma Tumor Syndromes (including Cowden syndrome, Lhermitte–Duclos disease and Bannayan–Riley–Ruvacalba syndrome) caused by mutations in PTEN; in all these cases the genetic defect leads to the hyperactivation of mTORC1. TSC and PTEN-mutated neurons have similar but not identical pathologic phenotypes, possibly reflecting subtle differences in the signalling that have yet to be discovered. Other neurodevelopmental disorders due to mTOR deregulation include epileptic encephalopathy, and neurodegenerative and psychiatric diseases.62 ,63 The present review underlines the role of mTOR hyperactivation in the major FE syndromes with or without MCDs (table 1). In particular, both germinal and somatic mutations have been reported in genes coding for different components of mTOR signalling, such as GATOR1 encoding genes, PIK3CA, AKT3 and MTOR itself.7 ,19 ,33 ,48 ,64 Despite several evidences implicating the deregulation of mTOR pathway in the pathogenesis of epilepsy, the way in which it triggers epileptogenesis is still unknown. mTORC1 hyperactivation could contribute to aberrant circuit formation or alter already established neural circuits. Furthermore, as mTOR activity is regulated by several non-genetic factors, a combination of mutations in mTOR signalling genes and environmental factors could hyperactivate mTOR synergistically, representing a possible explanation for the variable phenotypes found in mTORopathies.62 Moreover, MCD-related epilepsies due to mutations in mTOR pathway genes often show early onset and poor response to pharmacological treatment. This could be partly explained by the fact that anticonvulsant medications represent only a symptomatic therapy, which does not address the underlying biological mechanism. The implication of mTOR signalling in a wide variety of lesional and non-lesional FEs is now promoting the development of targeted therapies based on mTOR inhibitors, which could significantly improve patients' treatment and prognosis.65 ,66 In particular, the prototype drug for all mTOR inhibitors developed so far is rapamycin; some examples of available rapamycin analogues (rapalogs) are everolimus, temsirolimus and ridaforolimus.67 ,68 Second-generation mTOR inhibitors block the feedback activation of PI3K/AKT signalling acting as ATP-competitive mTOR kinase inhibitors; finally, drugs targeting downstream components of mTOR signalling pathway are being developed.68 ,69 Preclinical studies on animal models of TSC and other mTORopathies have shown a positive effect of rapamycin on the development of seizures and on seizure frequency, and early administration resulted also in a reduction of the alterations of cortical development. Moreover, clinical pharmacological trials on patients with TSC are now underway using rapalogs as everolimus, and first evidences support the potential role of this class of drugs,51 notwithstanding some concerns about the timing to obtain a curative effect and the possible associated side effects; the review by Citraro and colleagues70 comments on the last achievements of both preclinical and clinical trials using mTOR inhibitors to treat epilepsy or prevent epileptogenesis. Patients with refractory lesional epilepsy eligible for surgery, who carry mutations in mTOR pathway genes, represent a substantial challenge for the identification of the correct therapeutic strategy. For instance, it is still unknown whether germline mutations (which are present in every cell of the brain) underlie neural malfunctioning, which is more widespread than the focal lesion, as reported for TSC, where extended epileptogenic networks not restricted to tubers have been described.71 This implies that surgical resection of the detected EZ may not be sufficient to abolish seizures. Rare cases that underwent lesionectomy showed a good surgical outcome;40 however, more robust prognostic data are needed.
Conclusions
In this review, we outlined the role of GATOR1 mutations in FE. The involvement of DEPDC5, NPRL2 and NRPL3 genes in different lesional and non-lesional FEs is in contrast to the previous knowledge that mutations in specific genes are linked to epileptic syndromes in which seizures semiology suggested the main involvement of specific brain areas (as for LGI1 mutations in EAF, or neuronal nicotinic acetylcholine receptor in ADNFLE). The histological analysis of brain specimens in individuals with MCDs, as those found in TSC or GATOR1 mutated patients, shows the hyperactivation of mTOR pathway restricted to dysmorphic neurons, suggesting that even in those patients without a detectable MCD, the presence of a small group of dysmorphic neurons due to mTOR hyperactivation can elicit an epileptogenic effect. In those cases with a detectable MCD the excision of the EZ may represent a curative option, although the long-term outcome is still undefined. In pharmacoresistant patients, not eligible for surgery, the GATOR1/mTOR signalling represents a promising therapeutic target. Furthermore, as mTOR inhibitors have shown an anticonvulsant and an antiepileptogenic effect, this will open the way to a novel class of antiepileptic drugs that could reduce the neurological conditions derived by recurrent seizures when early administered to the patient.
References
Footnotes
Contributors SB wrote the first draft of the manuscript and designed the figures and table. All authors contributed to the critical reading and editing of the manuscript.
Funding We acknowledge Telethon Foundation (Grant GGP13200 to PT and TP) for financial support.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.