Article Text

Abstract
Mutations in the sodium-gated potassium channel subunit gene KCNT1 have recently emerged as a cause of several different epileptic disorders. This review describes the mutational and phenotypic spectrum associated with the gene and discusses the comorbidities found in patients, which include intellectual disability and psychiatric features. The gene may also be linked with cardiac disorders. KCNT1 missense mutations have been found in 39% of patients with the epileptic encephalopathy malignant migrating focal seizures of infancy (MMFSI), making it the most significant MMFSI disease-causing gene identified to date. Mutations in KCNT1 have also been described in eight unrelated cases of sporadic and familial autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE). These patients have a high frequency of associated intellectual disability and psychiatric features. Two mutations in KCNT1 have been associated with both ADNFLE and MMFSI, suggesting that the genotype–phenotype relationship for KCNT1 mutations is not straightforward. Mutations have also been described in several patients with infantile epileptic encephalopathies other than MMFSI. Notably, all mutations in KCNT1 described to date are missense mutations, and electrophysiological studies have shown that they result in increased potassium current. Together, these genetic and electrophysiological studies raise the possibility of delivering precision medicine by treating patients with KCNT1 mutations using drugs that alter the action of potassium channels to specifically target the biological effects of their disease-causing mutation. Such trials are now in progress. Better understanding of the mechanisms underlying KCNT1-related disease will produce further improvements in treatment of the associated severe seizure disorders.
- KCNT1
- Epilepsy and seizures
- ADNFLE
- MMFSI
- Potassium channel
Statistics from Altmetric.com
Introduction
Human KCNT1, which was first molecularly described in 2000,1 encodes a sodium-gated potassium channel subunit also known as Sequence Like a Calcium Activated K+ channel (SLACK), KCa4.1 or Slo2.2.2 The gene is highly expressed in the nervous system, and the encoded ion channel is thought to modulate hyperpolarisation following repetitive firing of action potentials.3 ,4 The first disease-causing mutations in KCNT1 were described in 2012 and were shown to cause two distinctly different types of epilepsy: the familial focal epilepsy syndrome autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE)5 and the epileptic encephalopathy malignant migrating focal seizures of infancy (MMFSI).6 The phenotypic spectrum associated with KCNT1 mutations has since expanded to include other forms of focal epilepsy and epileptic encephalopathies. KCNT1 mutations may also be associated with cardiac arrhythmias.
KCNT1 mutations in MMFSI
MMFSI, also known as epilepsy of infancy with migrating focal seizures, is an early-onset epileptic encephalopathy (EOEE) characterised by migrating multifocal seizures with onset before 6 months of age.7 Seizures are intractable to antiepileptic drugs, and patients experience severe psychomotor developmental delay.7 Barcia et al6 first identified de novo KCNT1 mutations in individuals with MMFSI using whole-exome sequencing (WES) and subsequently identified additional patients by direct Sanger sequencing. Subsequent studies using whole-exome, whole-genome or gene panel sequencing approaches in MMFSI and EOEE cohorts identified additional KCNT1 mutations in patients with MMFSI8–16 (figure 1 and table 1).
Clinical features of patients with KCNT1 mutations
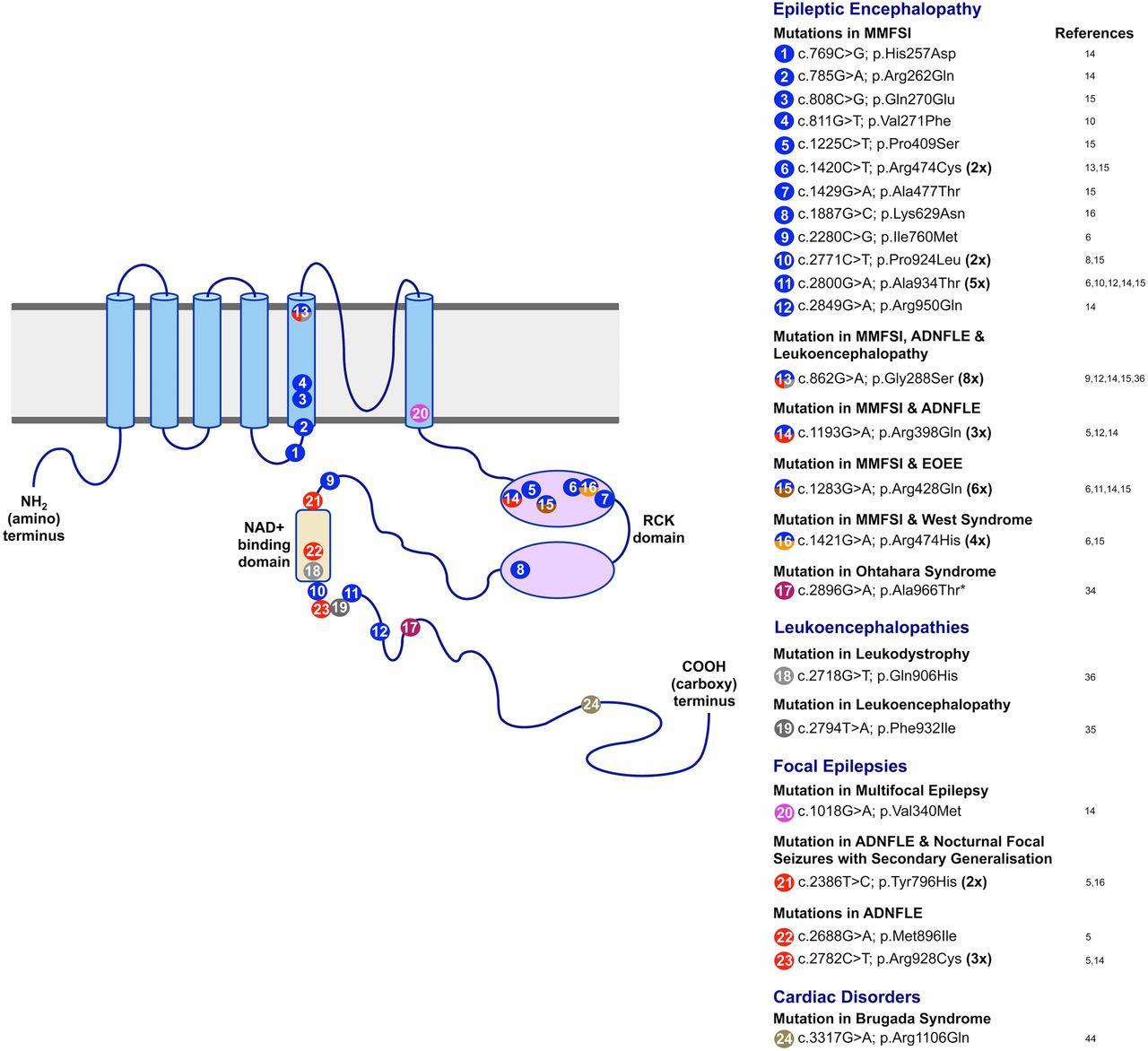
Diagram showing the KCNT1 protein structure and the positions of mutations identified in the gene to date. The KCNT1 protein consists of six hydrophobic transmembrane segments (S1–S6) with the pore-loop between S5 and S6. It has a large intracellular carboxy-terminal region containing tandem RCK domains and an NAD+ binding domain. The positions of missense mutations reported by previous studies are indicated, with different colours denoting different phenotypes. Mutations that are associated with more than one phenotype are marked with two or more colours. The numbers in bold type indicate the number of times recurrent mutations have been observed in multiple families and/or patients. ADNFLE, autosomal-dominant nocturnal frontal lobe epilepsy; EOEE, early-onset epileptic encephalopathy; MMFSI, malignant migrating focal seizures of infancy.
KCNT1 mutations have emerged as the most frequent genetic cause of MMFSI. Ten reports describing a total of 34 unrelated individuals with MMFSI with KCNT1 mutations have now been published6 ,8–16 (figure 1 and table 1). Twenty-four of these patients were confirmed to have de novo mutations, explaining why most cases occur in the absence of a family history. Importantly, parental somatic mosaicism of a KCNT1 mutation has been demonstrated in two cases,14 ,15 showing that there is a risk of recurrence of the disorder, where more than one child in a sibship could be affected, even if the parents are asymptomatic. Detection of parental somatic mosaicism of KCNT1 mutations is, therefore, important for genetic counselling of patients and their families.
To date, 39% (28 out of 71) of individuals from cohorts of patients with MMFSI that have been analysed for KCNT1 mutations are mutation positive.6 ,10 ,12 ,14 ,15 ,17 The high frequency of mutations is in striking contrast to the contribution of other genes associated with MMFSI since it was first described in 1995.7 Eight patients with MMFSI have been reported with mutations in SCN2A18 ,19 and three with missense mutations in SCN1A.20–22 Recessive mutations in TBC1D24, SLC25A22 and SLC12A5 have been described in four familial cases of MMFSI23–25 and four patients have been described with copy number mutations.20 ,26–28
KCNT1 mutations in ADNFLE
ADNFLE is a form of focal epilepsy in which patients experience violent motor seizures arising from sleep.29 The disorder has previously been associated with mutations in three nicotinic acetylcholine receptor (nAChR) subunit genes.30–33 Using a strategy combining whole-genome linkage analysis and WES in a large family with ADNFLE, Heron and colleagues5 identified a heterozygous missense mutation in KCNT1, p.Arg928Cys, which co-segregated with the epilepsy phenotype. Analysis of further patients identified KCNT1 missense mutations in two additional families with ADNFLE and a patient with nocturnal frontal lobe epilepsy (NFLE), the sporadic form of ADNFLE (figure 1). Patients with ADNFLE and NFLE with KCNT1 mutations were observed to have a more severe phenotype, which included an earlier age of seizure onset, a markedly increased frequency of refractory seizures and a high frequency of comorbid intellectual disability and psychiatric features compared with patients with ADNFLE with mutations in nAChR subunit genes5 (table 1). The majority of individuals with ADNFLE in two recent publications12 ,14 also exhibited intellectual disability and psychiatric features, including depression, attempted suicide, anxiety and attention-deficit hyperactivity disorder. KCNT1-associated ADNFLE often presents as a severe form of ADNFLE with the comorbidities being key features of the disorder. This observation should guide genetic testing.
Subsequent studies of patients with ADNFLE or NFLE have identified additional KCNT1 mutations and have shed some light on genotype–phenotype relationships. Møller and colleagues14 described a sporadic case of NFLE with the same heterozygous mutation as the original family with ADNFLE, p.Arg928Cys,5 highlighting that the same mutation may be seen in a familial or sporadic setting. A family with five individuals affected with either nocturnal focal epilepsy or MMFSI was found to have a heterozygous KCNT1 mutation, p.Arg398Gln.14 This mutation was previously seen in a family with ADNFLE.5 Similarly, Kim and colleagues12 identified a patient with ADNFLE with a heterozygous missense KCNT1 mutation p.Gly288Ser, and the same mutation has been reported in several unrelated patients with MMFSI.9 ,14 ,15 These findings are important as they demonstrate that the same KCNT1 mutation can give rise to either NFLE or MMFSI, even in members of the same family. The first reports of KCNT1 mutations5 ,6 gave rise to the idea that some mutations would cause ADNFLE while others would be responsible for MMFSI as different mutations were associated with each phenotype in these studies. More recent data, however, show this is not the case and it is now clear that genotype–phenotype correlations with KCNT1 mutations are not straightforward. While the identification of a KCNT1 mutation will aid in diagnosis and counselling of patients, providing a prognosis based on a molecular result is likely to remain challenging.
KCNT1 mutations in other seizure disorders
The identification of KCNT1 mutations in both ADNFLE and MMFSI, which are phenotypically distinct epileptic disorders, suggested that the two phenotypes may be part of a larger spectrum of epilepsies associated with mutations in the gene. This appears to be the case as KCNT1 mutations have subsequently been identified in patients with other forms of EOEE, focal epilepsy and leukoencephalopathies (figure 1 and table 1). In a Japanese cohort of 362 patients with EOEEs, a patient with West Syndrome and a patient with an unclassified EOEE were found to have de novo heterozygous KCNT1 mutations.15 An individual with Ohtahara syndrome was found to have a KCNT1 missense mutation, which was homozygous due to paternal isodisomy.34 Each of the three patients with EOEE showed severe drug-resistant epilepsy and developmental delay, similar to that of the MMFSI phenotype (table 1), and two showed delayed myelination. The range of focal epilepsy phenotypes associated with KCNT1 mutations was further expanded by the discovery of a heterozygous KCNT1 mutation p.Val340Met in a patient with multifocal epilepsy14 (figure 1 and table 1) and the identification of the p.Tyr796His mutation, previously reported in a family with ADNFLE, in a patient with severe nocturnal focal seizures with secondary generalisation and developmental regression.16 Heterozygous de novo KCNT1 mutations were identified using WES in a leukoencephalopathy patient with severe delayed myelination and refractory seizures35 and in two patients with hypomyelinating leukodystrophy, whose initial symptom was seizures in both cases.36 One of these was a novel mutation while the other was previously observed in both MMFSI and ADNFLE patients (p.Gly288Ser). These findings further demonstrate that KCNT1 mutations are highly pleiotropic and can cause a wide spectrum of seizure disorders.
Pleiotropy is often observed with genes associated with epilepsy. Other examples include mutations in SCN1A,37 KCNQ238 and GABRG2,39 which can cause both mild and severe epilepsy phenotypes. Several epilepsy genes are also associated with non-epileptic phenotypes. For example, mutations in PRRT2 can cause infantile epilepsy and/or paroxysmal movement disorders40 while mutations in TBC1D24 cause several forms of epilepsy and hearing loss as well as DOORS syndrome.41 In some cases, the pleiotropy results from the differing functional effects of particular mutations. In other cases, such as KCNT1, this explanation is unlikely as the same mutation is associated with different phenotypes, sometimes within one family. In these cases, the variation in phenotype is likely to be due to modifier genes, environmental effects or a combination of both.
KCNT1 mutational spectrum and penetrance
Adding to the variability in the phenotypic expression of KCNT1 mutations is incomplete penetrance. It originally appeared that KCNT1 mutations show 100% penetrance, invariably causing severe disease.5 ,6 More recent findings, however, indicate that this is not the case. One individual in a family with ADNFLE with a KCNT1 mutation did not have seizures and another individual from the same family was mildly affected, experiencing a single seizure.14 The father of the patient with Ohtahara syndrome described above, who was heterozygous for the mutation, was similarly unaffected.34
As mentioned above, parental somatic mosaicism of KCNT1 mutations has been demonstrated in two cases. A Japanese patient15 with the p.Pro924Leu mutation and an Australian patient14 with the p.Ala934Thr mutation inherited the mutations from their unaffected mothers who were confirmed to be somatic mosaic for the respective mutations. The levels of mosaicism were determined to be between 14% and 29% in samples of blood, saliva and nails from the mother of the Japanese patient15 and approximately 60% in blood in the mother of the Australian patient.14 These observations show that high levels of somatic mosaicism of KCNT1 mutations can exist without causing detectable seizure phenotypes.
Of the 24 different KCNT1 mutations described to date, nine have now been observed multiple (up to eight) times (figure 1). This suggests that mutational ‘hot-spots’ play a role in KCNT1-mediated pathogenesis. Seven of these recurrent mutations occur in CpG dinucleotides and are C>T or G>A changes. CpG dinucleotides are known to be sites of recurrent mutation due to the spontaneous deamination of 5-methylcytosine to thymine, resulting in G:T mismatches. The inaccurate repair of these results in mutation.42 ,43
The data reported to date show that there is considerable phenotypic variability and incomplete penetrance associated with KCNT1 mutations, with phenotypes ranging from individuals who are apparently unaffected, to ADNFLE, to devastating disorders such as MMFSI or other EOEEs. This cannot be attributed solely to differences in the effects stemming from the different amino acid alterations as phenotypic variability is seen associated with the same mutation even within genetically similar individuals within single families. Uncovering the biological basis of this variation in phenotype will improve our understanding of KCNT1 channel biology and our ability to diagnose and give accurate prognoses for patients with KCNT1 mutations.
KCNT1 mutations and possible links with cardiac disorders
A targeted resequencing study examining ion channel mutations in patients with the cardiac arrhythmia Brugada syndrome (Brs) identified a de novo heterozygous missense KCNT1 mutation, p.Arg1106Gln in one individual.44 The patient with Brs with the KCNT1 mutation was reported to have seizures, consistent with previous findings. Furthermore, one of the patients with ADNFLE in a family with a KCNT1 mutation reported by Møller and colleagues14 has cardiac arrhythmia with features resembling Brs, suggesting that there may be a link between perturbation of KCNT1 protein function and cardiac arrhythmias. Another member of the same family died due to sudden unexpected death in epilepsy (SUDEP), thought to be associated with cardiac dysfunction.14 These observations and the expression of KCNT1 in the heart1 ,45 may suggest involvement of KCNT1 in heart function, possibly in the regulation of action potentials similar to its role in the brain.
The possible association of KCNT1 mutations with cardiac disorders is part of an increasing body of evidence that both epilepsy and cardiac disorders can result from the same channelopathy. It is feasible that dysfunctional ion channels expressed in the brain and heart lead to the dysregulation of action potentials, contributing to both seizure activity and cardiac arrhythmia. Mutations in two known cardiac arrhythmia genes, SCN5A and KCNH2, have previously been found in patients with epilepsy. SCN5A mutations have been found in a patient with idiopathic epilepsy who died due to SUDEP,46 a family with Brs and epilepsy,47 and in a patient with neonatal seizures and long QT syndrome (LQTS).48 An increased frequency of seizure disorders was observed in patients with LQTS caused by mutations in KCNH249 and a KCNH2 mutation was found in a patient with a history of epilepsy who was undergoing cardiologic evaluation.50 These findings suggest that the association between genetic epilepsies and cardiac arrhythmias may be more significant than has previously been appreciated. The observation of cardiac disorders in patients with KCNT1 mutations and seizure disorders further supports the association of epileptic and cardiac channelopathies, and further studies are needed to shed light on the biology of this possible link.
Structure, expression and function of KCNT1 and its protein product
The human KCNT1 gene is located at chromosome 9q34.3 and comprises 31 exons. The gene is transcribed to produce a 4.7 kb mRNA that encodes a 138 kDa protein. Expression profiling of the human mRNA showed high expression in most brain regions, except the corpus callosum and substantia nigra.1
The full-length human KCNT1 protein contains 1235 amino acid residues and is the largest potassium channel subunit identified. It shares 76% amino acid identity (protein alignment: http://blast.ncbi.nlm.nih.gov) with and has been shown to interact with the KCNT2 protein (also known as SLICK),51 the only other protein in the sodium-activated K+ channel family.3 The KCNT1 protein comprises a small intracellular amino-terminal domain, six hydrophobic transmembrane segments (S1–S6) with a pore-loop between S5 and S6, and a large intracellular carboxy-terminal domain.3 ,52 The C-terminal domain contains tandem regulators of K+ conductance (RCK) domains and an NAD+ binding domain.3 ,53 The KCNT1 and KCNT2 subunits are thought to form a tetrameric channel51 that mediates a sodium-sensitive potassium current that is found in a range of neuronal cells.54–62 Its action of restoring polarisation after repetitive neuronal firing assists with the regulation of neuronal firing.63
The interaction of the KCNT1 and KCNT2 channel subunits raises the possibility that mutations in KCNT2 could cause similar phenotypes to those in KCNT1. However, no such mutations have been described in multiple exome sequencing studies of patients with epileptic encephalopathies.8 ,34 ,64–67 This strongly suggests that KCNT2 mutations do not cause epileptic encephalopathies with the same frequency as KCNT1 mutations. This may be because the expression patterns of the two genes do not completely overlap, suggesting some differences in their function. Differences in expression patterns of the two genes have been described in rodents.68 ,69 Data from the Allen Brain Atlas (http://human.brain-map.org) indicate that differing expression patterns of the two genes also occur in humans.
Two human KCNT1 transcripts have been identified to date (NCBI data: http://www.ncbi.nlm.nih.gov/gene/57582). In rodents, five distinct Kcnt1 transcripts have been identified empirically.4 These transcripts differ only at the 5′ end, producing subunits with differing N-terminal domains, but identical transmembrane and C-terminal domains. Two of the rodent transcripts are termed Slack-A and Slack-B. Studies in mouse and rat showed that these transcripts have differing expression patterns in the brain and produce proteins with different electrophysiological properties.4 ,52 ,70 ,71 Slack-A is rapidly activated following depolarisation while Slack-B has slower activation kinetics.4 Slack-A appears to have widespread expression while expression of Slack-B is restricted to the frontal cortex and some non-cortical regions.4 ,52 ,71
All studies of human KCNT1 thus far have focused on splice isoform 1 (NM_020822.2), which is partially orthologous to rodent Slack-A, but codes for a protein with additional sequence at the N-terminal end (figure 2). The expression of the transcripts orthologous to Slack-A and Slack-B in humans has not been empirically demonstrated; however, alignments of rat Slack-A and Slack-B protein to the KCNT1 genomic sequence in the Ensembl Genome Browser (http://www.ensembl.org) show that sequences homologous to the first and second exons specific for both Slack-A and Slack-B are present within the human KCNT1 gene (figure 2). It is, therefore, possible that the same isoforms with the same expression patterns exist in both humans and rodents.

Amino acid alignment for KCNT1 isoforms in human, rat and mouse. Amino acid sequences from the first and second exons of human KCNT1 isoform 1 (NM020822.2) were aligned to mouse and rat Slack-A and Slack-B isoforms previously reported by Brown et al.4 Amino acid sequences for the hypothetical human SLACK-A and human SLACK-B isoforms were derived from ENST00000486577 and ENST00000490355 respectively from the Ensembl Genome Browser.
Taken together, these studies show that KCNT1 is highly expressed in the brain. The gene in rodents appears to have several differentially expressed isoforms with different functional properties. The functional roles of these isoforms are not yet understood and their existence has not been empirically demonstrated in humans. Further study of the physiology and expression of the different isoforms is required before we can have a complete understanding of the differential forms of KCNT1 transcripts and the proteins they encode.
Effects of KCNT1 mutations on channel function
The 24 seizure disorder-related KCNT1 mutations described to date are all missense mutations, with no nonsense or other truncating mutations having been identified. This suggests that perturbation of normal KCNT1 protein function, rather than the loss of its function, underlies the pathogenic mechanism that leads to the observed phenotypes. This may be consistent with the phenotypes observed in a Kcnt1 null mouse model,70 which shows slightly reduced locomotive activity and impaired reverse learning memory and adaptation to new environments, but no seizures. Animal models based on KCNT1 mutations observed in human patients will be required in order to accurately analyse the mechanism of these disease-causing mutations.
The positions of KCNT1 mutations associated with seizure disorders show clustering in particular functional domains of the KCNT1 protein. Specifically, they alter amino acid residues located near to or within the S5 and S6 transmembrane segments, the RCK domains or the NAD+ binding domain (figure 1). The sole exception is the mutation seen in the patient with Brs, p.Arg1106Gln, which is located closer to the C-terminus and is not in a characterised functional domain. The S5 and S6 segments flank the pore loop of the channel, while both the RCK domain and the NAD+ binding domain allosterically regulate channel opening and potassium ion permeation.72–74 This suggests that most of the mutations found have the potential to alter channel opening.
In vitro functional studies using either the rat Slack-B variant6 ,12 ,34 or the human KCNT1 transcript 116 ,75 expressed in Xenopus oocytes (frog eggs) have shown that the KCNT1 mutations found in individuals with ADNFLE, MMFSI and Ohtahara syndrome result in an increase in current amplitude of between 3-fold and 22-fold compared with wild-type, indicating a hypermorphic mutational effect. Studies performed6 ,12 ,16 ,34 ,75 to characterise the electrophysiological effects of KCNT1 mutations have produced variable results in terms of the extent of increase in the current amplitude that is associated with particular mutations, and further characterisation is required. Additionally, patch-clamping experiments carried out in Xenopus oocytes demonstrated that channel cooperativity, that is, an increase in the time channels remain open for and the number of simultaneous channel openings, occurs when multiple (four or more) KCNT1 channels are present in the same patch.12 Cooperativity was increased in mutant channels, leading to a larger increase in channel current compared with wild-type than is seen with single channels. This cooperativity may also occur in vivo where channels are clustered in specific regions of neurons. Therefore, it is possible that studies done in whole Xenopus oocytes, as opposed to patch-clamping experiments, do not give a complete picture of the functional effects of endogenous KCNT1 mutations in neurons, as cooperativity is not observed in these experiments.
The KCNT1 channel subunit is involved in the slow hyperpolarisation after a single action potential or following repetitive firing of action potentials (figure 3).3 It has been speculated that in patients with KCNT1 mutations, enhanced potassium current in inhibitory interneurons leads to prolonged hyperpolarisation, resulting in an imbalance between neuronal excitation and inhibition that lead to seizures.12 ,75 The differing electrophysiological properties of the different KCNT1 isoforms may play a role in the expression of the different phenotypes caused by KCNT1 mutations. The disruption of the KCNT1 Slack-B-orthologue function may be the major cause of seizures in patients with ADNFLE, given its expression in the frontal cortex and its slow activation property that could facilitate the rhythmic firing of neurons4 ,71 that is likely to be required for the oscillatory currents observed during sleep (sleep spindles).76 Hypermorphic mutations in KCNT1 could lead to seizures in both MMSFI and ADNFLE due to an increase in Slack-A-orthologue function. Since Slack-A is rapidly activated following depolarisation,4 an increase in Slack-A current could lead to a more rapid membrane repolarisation, allowing faster repriming of sodium channels in excitatory neurons, resulting in a higher firing rate of action potentials throughout the brain. Additionally, it is possible that patients with MMFSI have ADNFLE-type seizures that are not observed due to the severity of neurological disease in MMFSI.

{kind=link}
{kind=link}
{kind=link}
Illustration of a general model of an action potential. As the membrane potential reaches threshold potential, voltage-gated sodium channels (eg, SCN1A) open, allowing sodium ions to flow into the cell. The membrane potential increases, and this stage is known as depolarisation. When membrane potential reaches +30 mV, sodium channels closes and potassium channels such as voltage-gated potassium channels (eg, KCNA1), voltage and calcium-sensitive potassium channels (eg, KCNMA1) and sodium-activated potassium channels (eg, KCNT1) begin to open, allowing the flow of potassium ions from the cell. With the potassium channels open, the membrane begins to repolarise back towards its resting potential. The closure for certain potassium channels such as KCNT1 is a delayed process, causing the membrane potential to fall below −70 mV, which is known as hyperpolarisation. The Na+/K+ pump restores the membrane potential from hyperpolarisation back to its resting potential. Figure adapted and modified from Bean.81
KCNT1 mutations may have effects additional to the demonstrated increase in potassium current. The C-terminal domain of the KCNT1 protein is known to interact with the Fragile X mental retardation protein (FMRP).77 Alterations in this interaction may be related to the comorbidities seen in patients with KCNT1 mutations. For example, it is possible that the high incidence of intellectual disability seen in patients with ADNFLE with KCNT1 mutations5 ,12 ,14 is due to altered interaction of the KCNT1 channel with FMRP.
Targeting KCNT1 mutations with quinidine
The identification of KCNT1 mutations as a genetic cause for MMFSI and ADNFLE suggests that these conditions may be treatable using a drug that specifically targets the KCNT1 channel. One such drug, quinidine, has recently garnered much interest. Quinidine is an inhibitor of several types of potassium channels, including KCNT1,78 ,79 and is currently used as an antiarrhythmic agent and antimalarial drug.80 Since functional studies have shown that mutations in KCNT1 cause an increase in the channel current,6 ,12 ,16 ,34 ,75 using an inhibitor to reverse this abnormal channel function could treat seizures in patients with hypermorphic mutations in KCNT1. Indeed, the effectiveness of quinidine in reversing the increased current of mutant KCNT1 channels has been demonstrated in vitro.16 ,75
Quinidine treatment has been trialled in three patients with KCNT1 mutations for whom treatment with standard antiepileptic therapies had been unsuccessful.11 ,16 Two patients with MMFSI showed marked improvement in seizure control following quinidine administration. One was seizure-free for 4 months at the time the trial was reported11 while the other had an 80% reduction in seizure frequency.16 Both patients’ developmental progress was improved, although the patient reported by Bearden and colleagues11 remained severely developmentally delayed, which was thought to be due either to irreversible brain damage caused by frequent seizures prior to quinidine dosing or developmental effects of the mutant KCNT1 protein, particularly its interaction with FMRP.77 The extent of improvement in the second patient's developmental progress was not described.16 The third patient, who had an unusual phenotype of nocturnal focal seizures with secondary generalisation and developmental regression, showed no significant improvement with quinidine treatment.16 The success of quinidine as an epilepsy treatment in two of three patients is encouraging for KCNT1 mutation-positive patients, who often have seizures resistant to multiple antiepileptic drugs. However, caution must be exercised when using quinidine as it acts on multiple potassium channels including those in the heart and could possibly cause a long QT interval and lead to cardiac arrest.11 As discussed above, cardiac arrhythmias were reported in a patient with ADNFLE and also in a patient with Brs carrying KCNT1 mutations, raising the possibility that undiagnosed cardiac disorders exist in patients with KCNT1 mutations.14 ,44 Hence, the required trials of quinidine in larger cohorts of patients with KCNT1 mutations should include ECG monitoring to examine potential cardiac side effects. Development of a specific KCNT1 channel-targeting inhibitor with good blood–brain barrier penetration may provide a safer and more effective alternative to quinidine.
Summary
Since the first report of mutations in KCNT1 by two groups in patients with epilepsy in 2012,5 ,6 a number of studies have confirmed the importance of the gene in human neurological disorders.8–16 ,34–36 Mutations in KCNT1 appear to cause a wide spectrum of seizure disorders, which include ADNFLE, multifocal epilepsy, MMFSI, Ohtahara syndrome and leukoencephalopathies. Moreover, KCNT1 mutations are being considered in disorders beyond neurological phenotypes and may be associated with Brs44 and other cardiac arrhythmias.14 Further studies are now required to understand the underlying biology of KCNT1-related disorders. This will improve the treatment of patients with KCNT1 mutations by enabling the use of therapies that target the specific biological pathways they affect. Indeed, work towards personalised medicine based on molecular genetic data has already begun with the trial of quinidine to treat patients with KCNT1 mutations.
References
Footnotes
Contributors CXL wrote the first draft of the manuscript and designed the figures. All authors contributed to the critical reading and editing of the manuscript.
Funding This work was funded by grants from the National Health and Medical Research Council of Australia: Career Development Fellowship 1032603 to LMD, Career Development Fellowship 1085984 to SEH and Project Grant 1070668 to SEH. CXL is supported by a President's Postgraduate Scholarship from the University of South Australia.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.