Article Text

Abstract
Background Neurodevelopmental disorders have challenged clinical genetics for decades, with over 700 genes implicated and many whose function remains unknown. The application of whole-exome sequencing is proving pivotal in closing the genotype/phenotype gap through the discovery of new genes and variants that help to unravel the pathogenic mechanisms driving neuropathogenesis. One such discovery includes TRIO, a gene recently implicated in neurodevelopmental delay. Trio is a Dbl family guanine nucleotide exchange factor (GEF) and a major regulator of neuronal development, controlling actin cytoskeleton dynamics by activating the GTPase Rac1.
Methods Whole-exome sequencing was undertaken on a family presenting with global developmental delay, microcephaly and mild dysmorphism. Father/daughter exome analysis was performed, followed by confirmatory Sanger sequencing and segregation analysis on four individuals. Three further patients were recruited through the deciphering developmental disorders (DDD) study. Functional studies were undertaken using patient-specific Trio protein mutations.
Results We identified a frameshift deletion in TRIO that segregated autosomal dominantly. By scrutinising data from DDD, we further identified three unrelated children with a similar phenotype who harboured de novo missense mutations in TRIO. Biochemical studies demonstrated that in three out of four families, the Trio mutations led to a markedly reduced Rac1 activation.
Conclusions We describe an inherited global developmental delay phenotype associated with a frameshift deletion in TRIO. Additionally, we identify pathogenic de novo missense mutations in TRIO associated with the same consistent phenotype, intellectual disability, microcephaly and dysmorphism with striking digital features. We further functionally validate the importance of the GEF domain in Trio protein function. Our study demonstrates how genomic technologies are yet again proving prolific in diagnosing and advancing the understanding of neurodevelopmental disorders.
- Microcephaly
- TRIO
- Dbl
- Rho GTPase
- Rac1
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Neurodevelopmental delay is genetically heterogenous and affects between 1% and 3% of the general population.1 The usage of next-generation genomic techniques such as whole-exome sequencing (WES) has facilitated the identification of numerous novel pathogenic variants implicated in the evolution of neurodevelopmental pathology.2 One such gene on chromosome 5, TRIO, has recently been identified as a candidate for human neurodevelopmental delay.3
Neurodevelopment is a complex process requiring coordinated axonal guidance and neuronal migration to facilitate the delivery of neurons from the neuroepithelium to their synaptic target. This process is driven by specific molecular cues, which regulate the neuronal growth cone, residing at the tip of the axon.4 Motility of the growth cone is, in part, regulated through remodelling of the actin cytoskeleton.5 ,6
Members of the Rho GTPase family are key modulators of cytoskeletal dynamics.7 They play an essential role in neurodevelopment through the control of actin cytoskeleton dynamics, including motility of the growth cone.8 Rho GTPases are activated by guanine nucleotide exchange factors (GEFs). TRIO encodes a GEF expressed in many tissues, including the central nervous system (CNS).9 ,10 It is highly expressed in many areas of the developing brain including the cerebellum, cortex, hippocampi and thalami.10 ,11 Interestingly, TRIO can be alternatively spliced, and, as a result, can encode several isoforms whose expression is nervous-system specific.11 ,12 Trio is thought to be one of the major regulators of neuronal development by controlling actin cytoskeleton remodelling through the activation of the GTPase Rac1.13 The importance of the Trio protein in neuronal development is further emphasised by the fact that total knockout or specific deletion of Trio in the mouse nervous system is embryonically lethal; embryos display defects in brain organisation and reduced brain size.9 ,14 Deletion of Trio, specifically in the hippocampus and in the cortex during early embryogenesis, results in aberrant organisation of these structures, impairing the learning ability of these mice.10
Our findings consolidate and extend the human phenotype recently reported by Ba et al.3 Through the application of parent/offspring WES analysis, we report three children from across the UK with microcephaly and neurodevelopmental delay who harbour de novo missense mutations in TRIO. We also report a family with TRIO mutations that segregate autosomal dominantly. We show for the first time that TRIO mutations identified in patients affect Trio function and consequently could be responsible for the phenotype observed.
Methods
Patients 1, 2 and 3 were assessed at the Wessex Clinical Genetics Service (figure 1). Exome sequencing of patients 2 and 3 was performed on DNA extracted from whole blood. Sequencing and analysis were undertaken as previously described;15 all variant positions reported are defined on GRCh37 (hg19) and TRIO transcript NM_007118. To validate the sample provenance, a parallel SNP panel was applied to confirm data identity.16
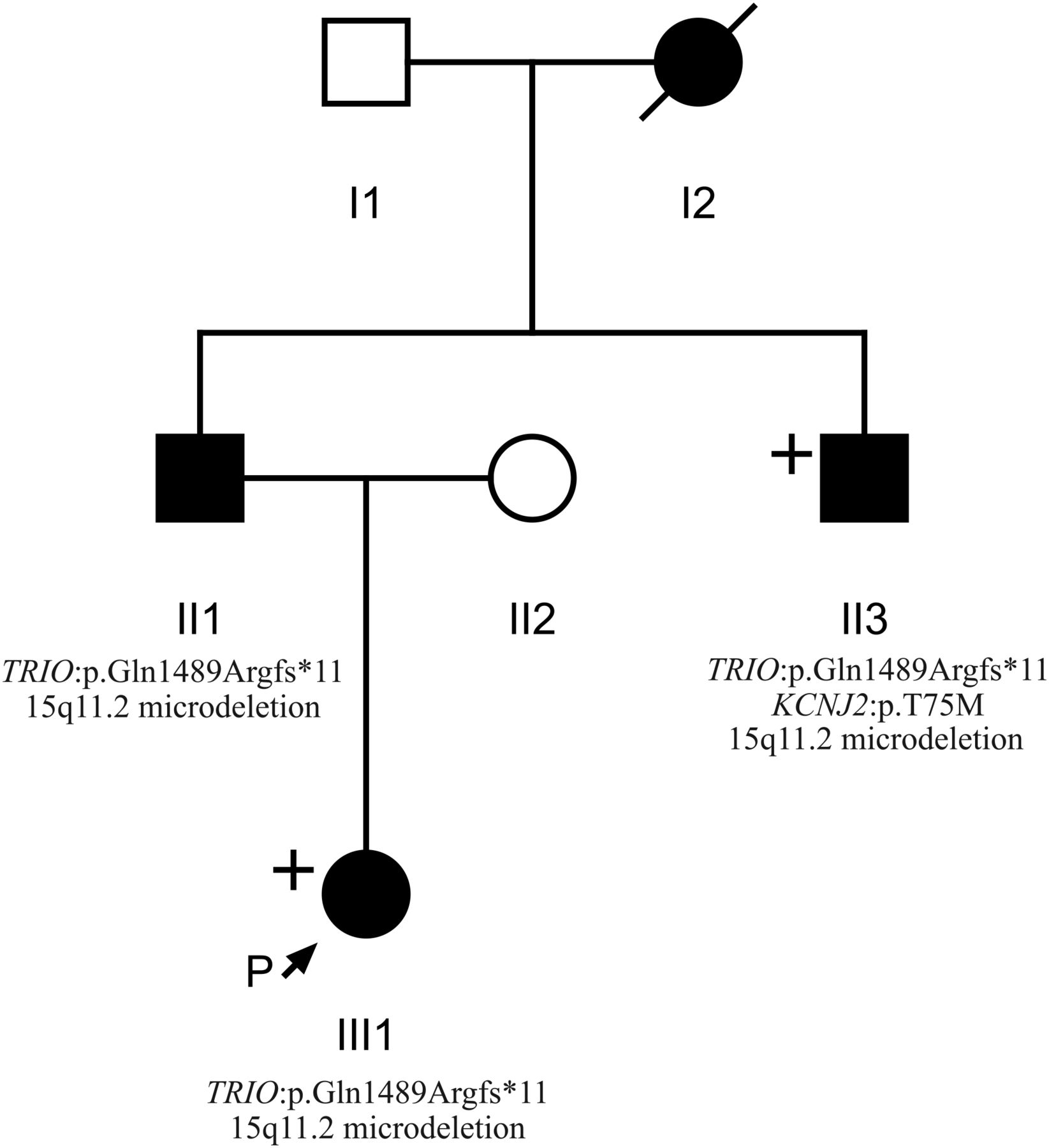
Pedigree of individuals 1, 2 and 3 (III1, II1 and II3, respectively) obtained through the Wessex Clinical Genetics Service. Affected individuals are shaded in black. Individuals who underwent whole-exome sequencing are labelled with a “+”. Genotypic information confirmed by Sanger sequencing is displayed where known.
Patients 4, 5 and 6 were referred to regional Clinical Genetics services across the UK, where they were recruited to the deciphering developmental disorders (DDD) study (http://www.ddduk.org). DDD has so far investigated over 4000 children with severe, undiagnosed developmental delay, and their parents, using a combination of genome-wide assays to detect all major classes of genetic variation in the protein-coding portion of the genome. Clinical information and phenotypes have been recorded using the Human Phenotype Ontology via a secure web portal within the DECIPHER database.17 ,18 DNA samples from patients and their parents were analysed by the Wellcome Trust Sanger Institute using high-resolution microarray analysis (array-comparative genomic hybridisation and SNP genotyping) to investigate CNVs in the affected child, and exome sequencing to investigate single-nucleotide variants and small insertions/deletions (indels). Putative de novo sequence variants were validated using targeted Sanger sequencing. The population prevalence (minor allele frequency) of each variant in nearly 15 000 samples from diverse populations was recorded, and the effect of each genomic variant was predicted using the Ensembl Variant Effect Predictor.19 Likely diagnostic variants in known developmental disorder genes were fed back to the referring clinical geneticists for external Sanger validation and discussion with the family via the patient’s record in DECIPHER, where they can be viewed in an interactive genome browser. Full genomic datasets were also deposited in the European Genome–Phenome Archive (http://www.ebi.ac.uk/ega).
Functional analysis to assess input of variants was undertaken. TRIO point mutants were generated by introducing a point mutant in the wild-type (wt) form of green fluorescent protein (GFP)-tagged Trio using the Quick change site-directed mutagenesis kit (Stratagene). The Rac1-guanosine triphosphate (GTP) pull-down assay was performed using the Cdc42/Rac1-interactive binding (CRIB) domain of PAK1 as described.20 Total HEK293T lysates and corresponding pull-downs retained on GST-Sepharose beads were processed for western blotting using the Rac1 (BD) and GFP (Clinisciences) antibodies.
Results
Patients 1, 2 and 3
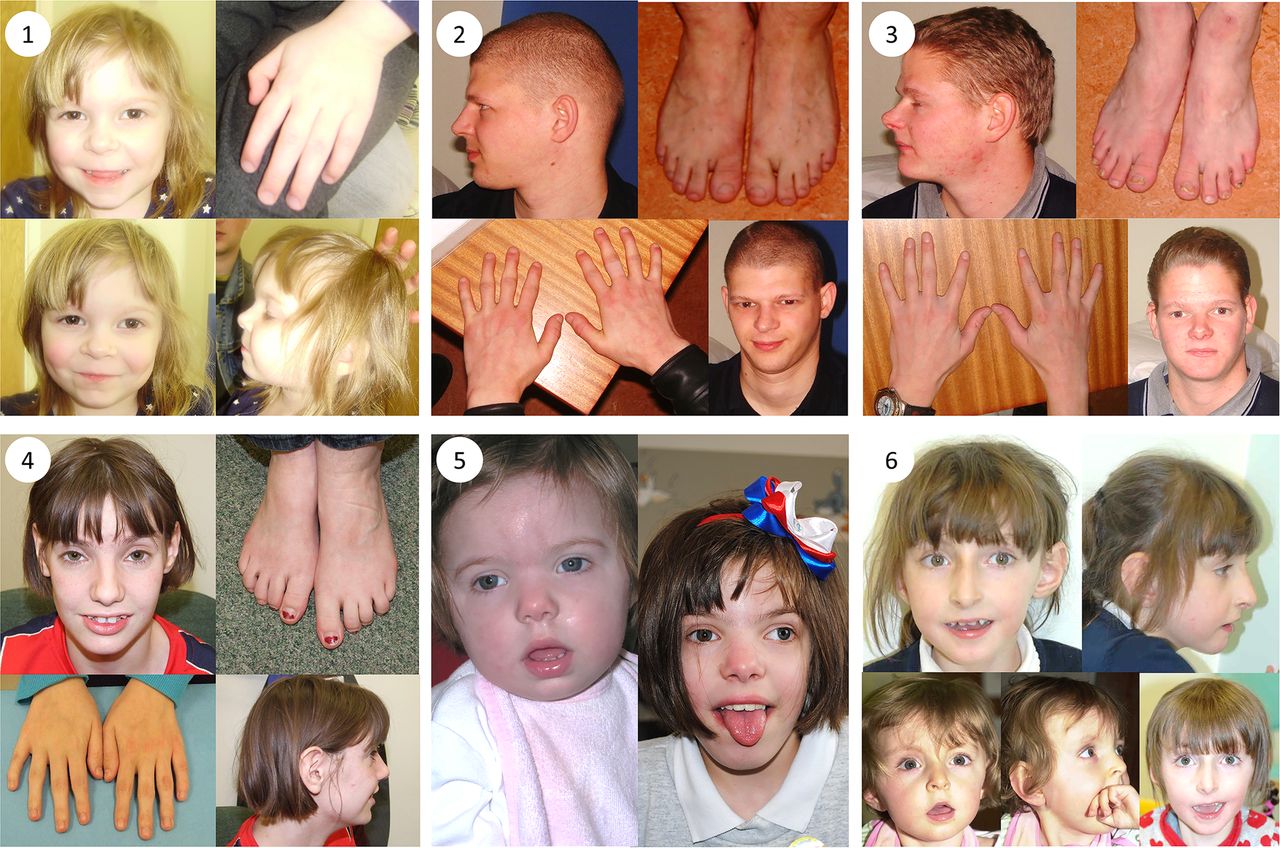
Patient 1 (see figure 1) was whole exome sequenced and a heterozygous frameshift deletion (p.Gln1489Argfs*11) in TRIO was identified, which was paternally inherited. This variant is predicted to cause nonsense-mediated decay of the transcript, and thus prevent expression of this allele (table 1).21 She was last assessed at the age of 17 months and was displaying signs of mild developmental delay; she sat unsupported at 9 months and walked unaided at 17 months. First words were spoken at 17 months and there was evidence of mild delay in expression and comprehension. She is a fussy eater and has dietetic input to increase calorific intake. There were some subtle behavioural signs including poor attention. She is microcephalic with a head circumference 5 SDs below the mean (figure 2). Dysmorphic features included a short nose, long philtrum, thin upper lip, epicanthic folds, 2/3 toe syndactyly and an almost absent fifth toe nail. She has right radial aplasia, a rudimentary thumb and absent first metacarpal. She was found to have multiple cardiac septal defects at birth; at the age of 3 she has two small apical muscular ventricular septal defects and a patent foramen ovale, none of which are of concern clinically. The pregnancy was complicated by pre-existing maternal type 2 diabetes mellitus, which required insulin therapy. She also has a 381 kb 15q11.2 microdeletion.
Clinical phenotypes
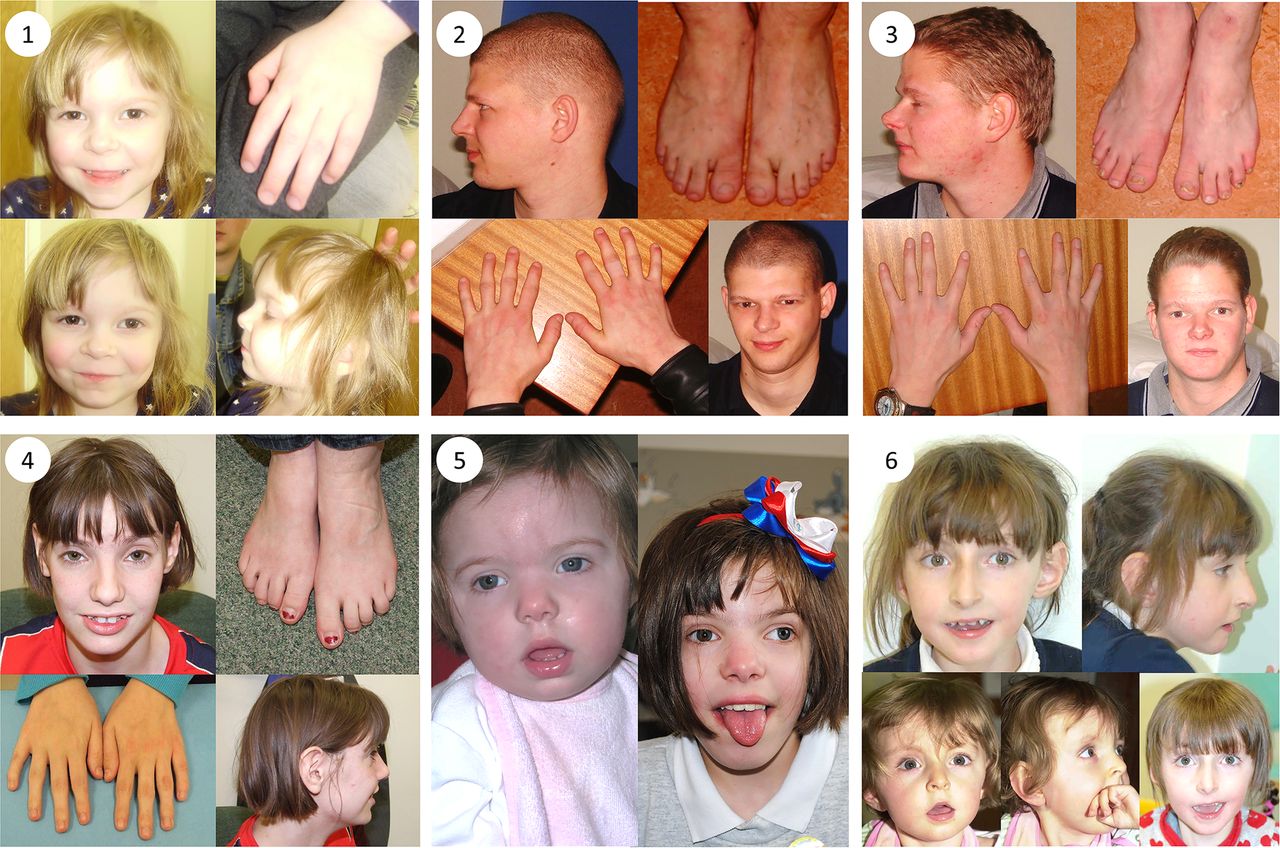
Clinical photographs. Photographs of individuals 1–6, taken at ages 3; 25; 22; 18; 2 and 11; 18 months, 7 and 10, respectively. All individuals harbour TRIO mutations. Individual 1 is the daughter of individual 2. Individuals 2 and 3 are brothers and have been previously described.32 Individuals 4, 5 and 6 are unrelated. Common features among studied individuals include microcephaly (seen in patients 1–5), mild dysmorphic facies, tapering fingers with prominent proximal interphalangeal joints, fifth finger clinodactyly and 2/3 toe syndactyly.
Patient 2 is the father of patient 1 and was genotyped using traditional Sanger methods. He harbours the same deletion in TRIO in addition to the same 15q11.2 microdeletion. He displayed mild learning difficulties in childhood and attended a specialist school. He is microcephalic with an occipital frontal circumference (OFC) 3 SDs below the mean. Other dysmorphic features include a straight nose, small jaw, pointed features, low anterior hairline, flattened thenar eminence, tapering digits, fifth finger clinodactyly and dental delay including absence or failed eruption of the lower central incisors and all four lateral molars.
Patient 3 is the uncle of patient 1 and brother of patient 2, who underwent WES. He harbours the same deletion in TRIO in addition to a pathogenic variant in KCNJ2 and the 15q11.2 microdeletion. He has learning difficulties and is microcephalic with a head circumference 5 SDs below the mean. He has similar dysmorphic features to his brother including a straight nose, small jaw, low anterior hairline, pointed features and short and tapering fingers. He has a structurally normal heart but experiences ventricular ectopic beats, which may be attributable to his coexisting variant in KCNJ2.22 Similar to his brother, he too has dental abnormalities with absence of any upper or lower lateral incisors. This family has been previously reported.23
Patient 4
Patient 4 has a de novo missense mutation in TRIO (p.Arg1428Gln). She was last assessed at the age of 16 years when she had evident global developmental delay. From a gross motor perspective, she sat unsupported at 10 months and walked at 22 months. It was not possible to elucidate the age of her first words; however, at the age of 16 years, although talkative, she remains unable to read or write. No feeding difficulties have been reported. This individual has numerous behavioural difficulties including stereotypies and obsessive compulsive traits. There was previous concern about aggressive and disruptive behaviour, which has now resolved. She is microcephalic with an OFC 5.41 SDs below the mean. Some dysmorphism was noted including congenital ptosis, upslanting palpebral fissures, large fleshy ears and fifth finger clinodactyly. Neurological features include insensitivity to pain and urinary incontinence.
Patient 5
Patient 5 was last assessed at the age of 8 years. She has a de novo missense mutation in TRIO (p.Pro1461Thr). She has global developmental delay and is microcephalic with an OFC 5 SDs below the mean. She sat unsupported at 11 months and walked unaided at 2½–3 years. Her first words were spoken between 4 and 5 years. Early feeding difficulties were reported including a poor suck, impaired bottle feeding and failure to thrive. Behavioural features include attention-deficit hyperactivity disorder and poor sleep.
Patient 6
Patient 6 has a de novo missense mutation in TRIO (p.Asn1080Ile) and she was last assessed at the age of 9 years. She has global developmental delay, most notably affecting gross motor and language development. She sat unsupported at 11 months and walked unaided at 4–5 years. She has no verbal communication and uses Makaton. She is still in nappies. There were some early feeding difficulties requiring gastrostomy; these have now resolved, although she remains with a thin body habitus. She has behavioural features that include hand stereotypies and aggressive episodes; she bites herself and can attack her younger brother. Head circumference is within the normal range. She has facial features that resemble Angelman-like facies with plagiocephaly. Neurological features include nocturnal tonic–clonic seizures, and a wide-based and ataxic gait.
TRIO mutations
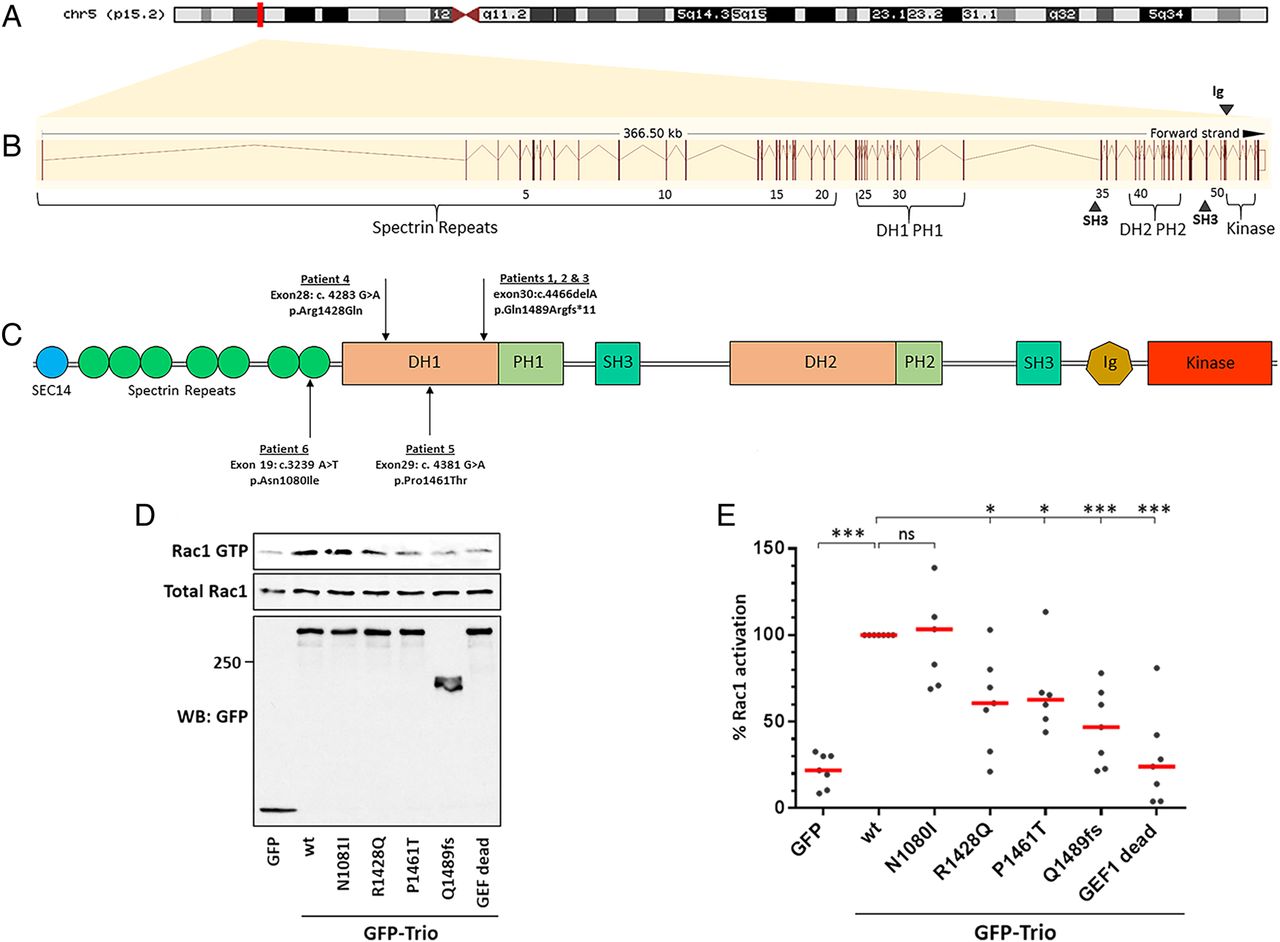
The six patients presented in this study have mutations in TRIO, a gene containing 57 exons that encodes a member of the Dbl family of GEFs, displaying strong conservation across evolution.13 Alternative splicing of TRIO results in isoforms that harbour either one or both of the highly conserved Dbl homology–Pleckstrin homology (DH–PH) domains. Out of the six Trio isoforms, five contain the first DH–PH domain, which specifically catalyses exchange of GDP for GTP on RhoG and Rac1.
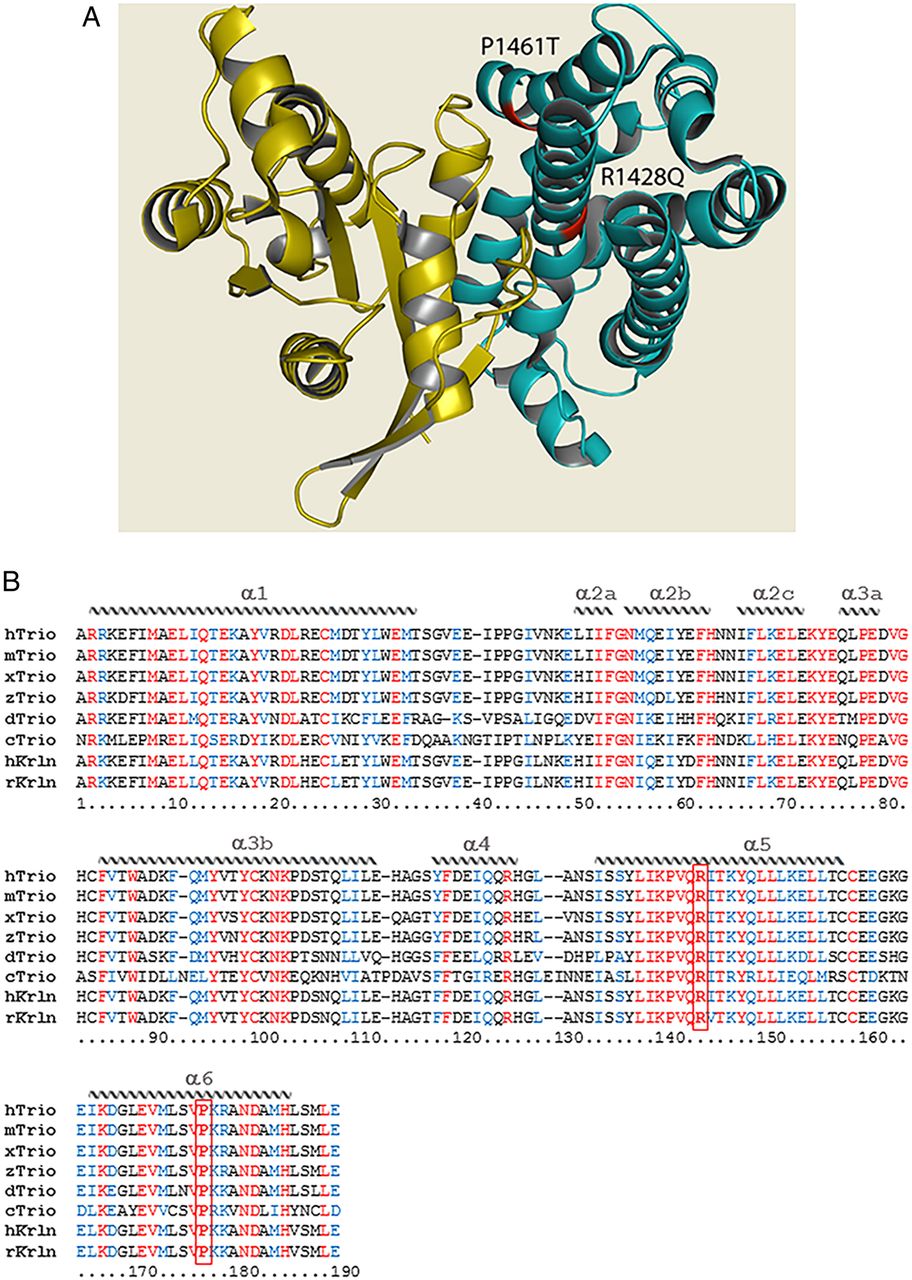
We set out to test whether the mutations identified in Trio could impact on its function and consequently contribute to the phenotypes observed in the six patients. The RhoGEF Trio is a major regulator of neuronal development, mainly through its activation of Rac1. Patients 1, 2 and 3 share a frameshift mutation (p.Q1489fs*) resulting in a truncated Trio protein (figure 3); it would be expected that this truncated transcript would be degraded by nonsense-mediated decay, resulting in the production of negligible protein product. Patients 4, 5 and 6 harbour de novo missense mutations. Interestingly, among the four mutations identified, three lie within the GEFD1 domain, responsible for Rac1 activation (p.R1428Q, p.P1461T and p.Q1489fs*) (figure 3C). Furthermore, the mutations affect highly conserved residues, since both R1428 and P1461 are present in Trio from invertebrates to mammals (figure 4B). They lie, respectively, within the α-5 and α-6 helices of the DH domain, which have been shown to interact with the target GTPase and occur at the protein–protein interface (figure 4A).24–26 The truncated mutant of Trio, p.Q1489fs*, is missing the PH1 domain, which is shown to be required for efficient GDP/GTP exchange.27 In contrast to the other five patients, individual 6 has a missense mutation (p.N1080I) within a spectrin repeat (figure 3C).

TRIO and its functional domains. (A) Genomic location of TRIO on the short arm of chromosome 5p15.2. (B) Graphical representation of the TRIO gene, comprising its 57 exons (vertical lines) spanning 366.5 kb. Exons are numbered alongside their relative position to coding domains. (C) Schematic overview of the Trio protein and its domains alongside the four mutations identified in the six patients from this study. Trio displays three enzymatic domains. Each guanine nucleotide exchange factor (GEF) module contains a catalytic domain, called Dbl homology (DH) domain (in reference to Dbl, the first RhoGEF identified as an oncogene in mammalian cells), and a Pleckstrin homology (PH) domain that plays a role in GEF activation and localisation. The first GEF domain, GEFD1, activates Rac1 and RhoG. The second GEF domain, GEFD2, acts on RhoA. In addition, Trio harbours numerous accessory domains. Listed from the N-terminus to the C-terminus, these include a CRAL-Trio/Sec14 motif; several spectrin-like repeats; two Src-homology 3 (SH3) motifs; and an immunoglobulin (Ig)-like domain. (D) Rac1-GTP pull-down assay. HEK293T cells (transfected as indicated) were lysed and active GTP-Rac1 was affinity purified using the Cdc42/Rac1-interactive binding (CRIB) domain of PAK1, immobilised on Glutathione-Sepharose beads. Purified GTP-bound and total Rac1 were detected by western blot, using an anti-Rac1 antibody. Protein expression in the cell lysates was verified by immunoblotting with an anti-green fluorescent protein (GFP) antibody. One representative experiment is shown. (E) Quantification of the Rac1-GTP pull-down assay shown in (D). Rac1 activation mediated by wt Trio was arbitrarily set to 100%, in order to be able to compare the individual experiments. The % of Rac activation was calculated from at least six independent experiments (mean ±SEM). *p< 0.015, ***p< 0.0001. Of note, all three mutations lying in the DH1 domain strongly affect Trio-mediated Rac1 activation.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The structure of Trio. (A) Quaternary structure of the Dbl homology (DH) domain of Trio (teal) in complex with the small GTPase substrate (yellow). Mutations within the DH seen in patients 4 and 5 are indicated in red; both mutations can be seen to occur at the protein–substrate interface. PDB IDs: DH domain—1NTY; GTPase substrate—1KZ7.33 ,34 Figure generated using PyMOL.35 (B) Sequence alignment of the DH1 domain of Trio (and Kalirin) across evolution. Sequences were obtained from NCBI databases and aligned with Clustal Omega. Identical residues are labelled in red, similar residues in blue. The structural features of the protein domain (α-helices) are depicted schematically and labelled on top of the sequence. The positions of the mutations p.R1428Q and p.P1461T are indicated in bold and boxed in red. Note that p.R1428Q and p.P1461T each affect a highly conserved residue within a very conserved region of the DH domain, in helices α-5 and α-6. Helices α-1, α-3b, α-5 and α-6 make contact with the target GTPase. Represented species are Homo sapiens (h), Mus musculus (m), Rattus norvegicus (r), Xenopus laevis (x), Danio rerio (z), Drosophila melanogaster (d), Caenorhabditis elegans (c).
We tested whether the mutations identified in our six patients affected Trio-mediated Rac1 activation (figure 3D,E). We introduced four mutations individually into the Trio cDNA and expressed wt or mutant GFP-tagged Trio proteins in HEK293T cells. Rac1-GTP pull-down experiments were performed using the CRIB domain of PAK1. As expected, Rac1-GTP was efficiently pulled down from cells expressing wt Trio. In contrast, the pull-down of active Rac1 from cells expressing the Trio p.R1428Q, p.P1461T or the truncated mutant p.Q1489fs* was strongly reduced, although to a lesser extent than the fully dead GEF mutant used as a negative control (Q1427A/L1435E).28 The results obtained with the two point mutants in the GEFD1 domain are therefore consistent with a crucial function of these two residues in Rac1 activation. The p.N1080I mutant (in individual 6) did not affect Trio-mediated Rac1 activation. Taken together, our results reveal that in five out of six patients harbouring a mutation in the TRIO gene, Rac1 activation is strongly reduced, demonstrating that Trio function is affected in these patients.
Discussion
TRIO is highly expressed in the developing brain; rat models show transcripts present in all major brain areas postnatally.11 ,29 TRIO and its paralog kalirin play a fundamental role in mammalian neuronal development. Trio deletion in mice results in impaired neuronal migration and axon guidance defects, global brain reduction and aberrant structural development of the hippocampus, cerebellum and cortex.9 ,14 Complete knockout mice are embryonically lethal; they have smaller brains and aberrant organisation in the hippocampus and hindbrain.9 ,10 ,14 A mouse model recently reported with a Trio knockout specific to the hippocampus and brain cortex addressed the role of Trio in postnatal brain development, concluding that Trio regulates the neuronal development of the hippocampus and affects the learning ability and intelligence of adult mice.10
Five of the six patients we report share a core phenotype comprising intellectual disability (ID), behavioural difficulties, subtle dysmorphism and microcephaly. This presentation aligns with the phenotype recently presented by Ba et al,3 who screened 2300 ID cases and identified four truncating mutations in TRIO. Similarly, they described skeletal anomalies, short stature, feeding difficulties and facial asymmetry. Perhaps most striking, however, are the similarities of the digital dysmorphology, most notably phalangeal hypoplasia, swollen proximal interphalangeal (PIP) joints and tapering fingers shared between their study and our findings. The microcephalic presentation is likely to be primary in nature, in which the skull is small because the underlying brain is small. Noteworthy, the microcephaly we describe is more severe than in the individuals reported by Ba et al,3 who all harboured loss-of-function mutations. Of interest, we showed that microcephaly was only present in individuals who harboured mutations in the GEFD1 domain affecting Rac1 activation by Trio. Since Trio function in the nervous system is mainly mediated by Rac1 activation, we can propose from these data that alteration of Rac1 activation by Trio variants is associated with microcephaly. Furthermore, we have recently shown that Trio is implicated in cell division by counteracting the effect of MgcRacGAP on Rac1 during cytokinesis.30 Interestingly, many genes associated with cell-cycle regulation have been found to be mutated in different forms of microcephaly.31 ,32 Therefore, the microcephaly phenotype observed in patients with TRIO mutations could also originate from a cell-cycle defect.
In contrast to patients 1–5, one individual in our study (individual 6) is somewhat of a phenotypic outlier; although she has ID, behavioural difficulties and dysmorphism, she is the only patient we report without microcephaly and who has seizures, complete speech failure and plagiocephaly. Unlike individuals 1–5 who have mutations affecting the DH1–PH1 domain, individual 6 has a missense mutation that affects a spectrin repeat and was functionally shown not to affect Trio-mediated Rac1 activation (figure 3). Spectrin repeats are important for Trio function as they bind many Trio regulators, such as NAV1, DISC1 and KidIns220.13 ,33 Therefore, we can hypothesise that the mutant Trio, p.N1080I, is mediated by a pathway independent of Rac1 activation, and could impair the binding of Trio to its interactors, potentially leading to protein mislocalisation.
TRIO is expressed in many tissue types; however, its role outside of the CNS remains to be fully elucidated. In addition, interpretation of a truly specific TRIO phenotype is confounded by additional findings; notably a 15q11.2 microdeletion in patients 1 and 2 and a pathogenic KCNJ2 variant in patient 3. Mutations in KCNJ2 have been associated with Anderson–Tawil syndrome, short QT and familial atrial fibrillation,22 ,34 likely explaining the ventricular ectopic beats seen in individual 3. It is also of note that individual 1 has right radial aplasia and ventricular septal defects, which may be secondary to an in utero environment complicated by maternal type 2 diabetes.35
Two of our probands displayed additional neurological signs including seizures, ataxia and pain insensitivity, and three of the probands had feeding difficulties. Furthermore, the ID reported was variable. The phenotypic variability could be related to the differing variant types (loss of function vs missense) and the importance of mutations affecting different exon positions within the DH1–PH1 domain and their consequent effect on protein function. Of particular interest, there was little difference in phenotypic severity between individuals harbouring loss-of-function mutations versus missense mutations that did not significantly alter protein length, and nor in the biological disruption of Trio-mediated Rac1 activity.
Conclusions
We are the first study to describe pathogenic missense mutations in TRIO and a TRIO syndrome that segregates in an autosomal-dominant pattern. Our findings corroborate and extrapolate previous literature; we show that mutations in TRIO affecting the RhoGEF1 domain cause a phenotype comprising microcephaly, ID, behavioural difficulties and dysmorphism with specific digital features. We functionally prove that genotypic, patient-specific TRIO mutants cause protein dysfunction modulated through reduced Rac1 activity, and that microcephaly was only present in individuals who harboured mutations in the GEFD1 domain. It now remains to be established whether the phenotypic contribution of TRIO variants in patients are attributable to the function of Trio in cell division and/or neuronal differentiation.
Acknowledgments
Thank you to all the patients and their families for participating in this study. We further extend our gratitude to the DDD study. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund [grant number HICF-1009-003], a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute [grant number WT098051]. The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
References
Footnotes
RJP, SG-H, SS, EGS, SE and DB contributed equally.
Contributors DB conceived the project and coordinated cases and functional work, contributed to the writing, examined and phenotyped the cases, edited the manuscript and contributed funding for sequencing. SE, RJP, MRJ and EGS undertook WES of the large family and discovered the first TRIO mutations. They jointly wrote and edited drafts. SGM, MJP and DG all contributed patients and their phenotypes through the DDD Study who did the WES of these three cases. SG-H and CM are clinicians involved in the care and phenotype of the large dominant family. CF-K, SS and AD performed the functional assays.
Funding RJP is supported by the University of Southampton. The functional work was supported by an Agence Nationale de la Recherche grant (ANR-14-CE11-0025-01) awarded to AD.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC).
Provenance and peer review Not commissioned; externally peer reviewed.