Article Text

Abstract
Background Joubert syndrome (JS) is a recessive ciliopathy characterised by a distinctive brain malformation ‘the molar tooth sign’. Mutations in >27 genes cause JS, and mutations in 12 of these genes also cause Meckel-Gruber syndrome (MKS). The goals of this work are to describe the clinical features of MKS1-related JS and determine whether disease causing MKS1 mutations affect cellular phenotypes such as cilium number, length and protein content as potential mechanisms underlying JS.
Methods We measured cilium number, length and protein content (ARL13B and INPP5E) by immunofluorescence in fibroblasts from individuals with MKS1-related JS and in a three-dimensional (3D) spheroid rescue assay to test the effects of disease-related MKS1 mutations.
Results We report MKS1 mutations (eight of them previously unreported) in nine individuals with JS. A minority of the individuals with MKS1-related JS have MKS features. In contrast to the truncating mutations associated with MKS, all of the individuals with MKS1-related JS carry ≥1 non-truncating mutation. Fibroblasts from individuals with MKS1-related JS make normal or fewer cilia than control fibroblasts, their cilia are more variable in length than controls, and show decreased ciliary ARL13B and INPP5E. Additionally, MKS1 mutant alleles have similar effects in 3D spheroids.
Conclusions MKS1 functions in the transition zone at the base of the cilium to regulate ciliary INPP5E content, through an ARL13B-dependent mechanism. Mutations in INPP5E also cause JS, so our findings in patient fibroblasts support the notion that loss of INPP5E function, due to either mutation or mislocalisation, is a key mechanism underlying JS, downstream of MKS1 and ARL13B.
- Genetics
- Cell biology
Statistics from Altmetric.com
Introduction
Human ciliopathies embody a rapidly growing group of disorders characterised by dysfunction of the primary cilium, a membrane-bound bundle of microtubules that projects from the apical surface of most cells.1 In addition to transducing chemosensation, mechanosensation and/or light sensation depending on the cell type, primary cilia mediate, among others, sonic hedgehog, Wnt, Hippo, PDGFα and G-protein coupled receptor signalling. Dysfunction of primary cilia results in a spectrum of phenotypes including central nervous system malformations, retinal dystrophy, cystic renal disease and hepatic fibrosis.2
Joubert syndrome (JS; MIM# 213300) and Meckel syndrome (MKS; MIM# 249000) are two recessive ciliopathies with overlapping phenotypic features. The defining feature of JS is the molar tooth sign (MTS) on brain MRI: cerebellar vermis hypoplasia, thick, elongated and horizontally oriented superior cerebellar peduncles, and a deep interpeduncular fossa.3 Clinically, JS is characterised by cognitive impairment, hypotonia, ataxia, abnormal eye movements, and episodic apnoea and/or tachypnoea in the neonatal period.4 Variable additional features have been observed, including other central nervous system anomalies (agenesis of the corpus callosum, polymicrogyria, heterotopia and occipital encephalocele), chorioretinal coloboma, retinal dystrophy, cystic renal disease, hepatic fibrosis and polydactyly.5–16
MKS is characterised by a posterior fossa brain malformation (typically occipital encephalocele), cystic renal disease, congenital hepatic fibrosis (eg, ductal plate malformation) and postaxial polydactyly.17 ,18 Phenotypic variability is also present, and other characteristics can include microphthalmia, situs inversus, skeletal abnormalities and Dandy-Walker malformation.19 ,20 Whereas individuals with JS typically survive beyond infancy, MKS is usually lethal in the fetal or neonatal period.
To date, mutations in at least 27 genes have been shown to cause JS, including NPHP1, AHI1, CEP290, RPGRIP1L, TMEM67, ARL13B, CC2D2A, INPP5E, OFD1, TMEM216, TCTN1, TCTN2, KIF7, TMEM237, CEP41, TMEM138, TMEM231, C5ORF42, TCTN3, IFT172, PDE6D, MKS1, CSPP1, B9D1, B9D2, C2CD3 and CEP120.21–25 Mutations in at least 13 genes have been shown to cause MKS, including MKS1, TMEM216 (MKS2), TMEM67 (MKS3), CEP290 (MKS4), RPGRIP1L (MKS5), CC2D2A (MKS6), NPHP3 (MKS7), TCTN2 (MKS8), B9D1 (MKS9), B9D2 (MKS10), TMEM231 (MKS11), TCTN3 and CSPP1.23 ,26 Different mutations in at least 12 of these genes, can cause either JS or MKS, supporting the notion that JS and MKS represent mild and severe presentations of the same biological disorder. Due to the genetic overlap between JS and MKS,22 ,27 ,28 we evaluated a large cohort of individuals with JS for mutations in MKS1.
Most of the proteins encoded by genes involved in JS and MKS localise to a structure at the proximal part of the cilium called the transition zone (TZ).28 ,29 The TZ anchors the cilium to the plasma membrane, and restricts and facilitates the movement of proteins in and out of the cilium.29 ,30 A few of the JS genes, among others ARL13B,31 INPP5E,32 CSPP133 and IFT172,34 encode proteins that localise to the cilium. Ciliary localisation of ARL13B depends on TZ function, while ciliary localisation of INPP5E depends on ARL13B, CEP164 and PDE6D function.35–37 Therefore, INPP5E dysfunction (due to mutation or mislocalisation) is likely to be key to the JS disease mechanism.
Similar to a recent report of two individuals with JS with biallelic MKS1 mutations,22 we identify mutations in MKS1 as the cause of JS in nine families, supporting the notion of genetic overlap between JS and MKS. These mutations (eight of them previously unreported) are associated with variable defects in cilium length and number in patient fibroblasts, but a consistent decrease in ciliary localisation of INPP5E and ARL13B, confirming in vivo and in vitro studies showing that INPP5E localisation, likely through effects on ARL13B localisation, is a central molecular defect underlying JS development.
Methods
Participants
Participants were enrolled under approved human subjects research protocols at the University of Washington, Seattle Children's Hospital, and the University of Utrecht, Wilhelmina Children's Hospital, the Netherlands. All participants or their legal guardians provided written informed consent. Inclusion criteria were: (1) MTS on brain imaging (or cerebellar vermis hypoplasia on CT scan when an MRI was not available) and (2) clinical findings of JS (intellectual impairment, hypotonia, ataxia).
Clinical and imaging data
Clinical information was collected using a structured intake form and review of medical records. At the time of enrolment, we reviewed brain MRI and/or CT scans to confirm the MTS and to evaluate for other structural brain abnormalities. When MRI or CT images were not available, we abstracted information from the MRI or CT report.
Mutation identification
Samples from participants at the University of Washington were sequenced using a modified molecular inversion probe capture method, followed by sequencing on an Illumina HiSeq.38 Exons and consensus splice sites (±2 bps) were targeted, and samples were considered sequenced if >80% of the targeted bps had >25X coverage. Samples from Utrecht University Medical Center were sequenced for 621 ciliary genes including the known Joubert genes and MKS1 (NM_017777.3). Deep sequencing was performed on two pooled sample cohorts of 32 and 34 cases, of which 51 cases had a diagnosis of JS. Sixty nucleotide long probes uniquely mapping to coding sequences of the 621 ciliary genes from the GRCh37/hg19 human reference genome with 50 bp flanks into intronic regions were designed with an average tiling density of 4 bp on average for positive and negative strands. The size of the targeted region was 2.7 Mb, covered by 779592 probes. Fragment library preparation and genomic enrichment on a 1 M custom microarray (Agilent Technologies, California, USA) were performed as previously described.39 The pooled samples were run as a full slide on the SOLiD 5500XL. Following SOLiD sequencing, colour space reads were mapped against GRCH37/hg19 reference genome using a custom pipeline based on the BWA software, and variants and small indels were annotated as described previously.39 Average sample coverage was 147X and 136X, and 92% and 89% of requested sequences were covered by more than 20 reads for run 1 and run 2, respectively.
Controls
The frequency of missense variants in subjects without severe congenital disorders was examined using data available through the NHLBI Exome Sequencing Project (ESP), Seattle, WA.40 For the p.S372del variant, we evaluated 182 samples from neurologically normal European American individuals by Sanger sequencing.
Cell culture
Retinal pigment epithelial cells and murine inner medullary collecting duct cells (IMCD3) were cultured in Dulbecco's modified Eagle's medium:F12 (1:1) (GlutaMAX, GIBCO), supplemented with 10% fetal calf serum and penicillin and streptomycin. Human fibroblasts were grown from skin biopsies in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and penicillin and streptomycin. Cells were incubated at 37°C in 5% CO2 to approximately 90% confluence. Fibroblasts were serum starved for 48 h prior to fixation. Details on MKS fibroblasts (MKS-158) can be found in online supplementary table S1 (Subject: Khaddour ‘07:562). ARL13B-277 fibroblasts have het c.246G>A (p.W82*) and het c.598C>T (p.R200C) mutations.31 INPP5E-171 fibroblasts have hom c.956G>A (p.G286R) mutations.41 ARL13B-277 and INPP5E-171 do not have mutations in other known JS-related genes.
In vitro mutagenesis
A human cDNA expression construct for MKS1 in a pCMV6-XL5 vector was ordered from Origene (SC123690; not full-length) and disease-associated mutations were introduced using site-directed mutagenesis (QuikChange II, Agilent) and sequence verified using Sanger sequencing (primers available upon request).
Transfection
Cells were seeded for at least 16 h prior to transfection with lipofectamine 2000 (Invitrogen, 11668-019) with Opti-MEM (Invitrogen, 31985-062) diluted DNA expression constructs, according to the supplier's protocol. After replating, cells were transfected with Lipofectamine RNAimax (Invitrogen, 13778-075) with ON-TARGETplus siRNA SMARTpools (Thermo Scientific Dharmacon): Non-targeting pool (D-001810-10) or Mks1 (L-063962-01), according to the supplier's protocol.
RT-qPCR
RNA was isolated (RNeasy Mini Kit, QIAGEN, 74106) and measured (NanoDrop spectrophotometer ND-1000, Thermo Fischer Scientific Inc.). cDNA was synthesised using the iScript cDNA Synthesis Kit (Bio-Rad, 170-8891) according to the supplier's protocol. RT-qPCR determined expression of Mks1, normalised against reference gene Rpl27. The primers (Sigma) used: mMks1 forward 5′-GGAGGTTCTTCATTGGCG-3′, mMks1 reverse 5′-TTGTCTCAGTGCGGAATCC-3′, mRpl27 forward 5′-CGCCCTCCTTTCCTTTCTGC and mRpl27 reverse 5′-GGTGCCATCGTCAATGTTCTTC. Samples were run with iQ SYBR Green Supermix (Bio-Rad, 170-8880) and CFX96 Touch Real-Time PCR Detection System (Bio-Rad); 95°C for 3 min, followed by 40 cycles of 10 s at 95°C, 30 s at 60/53°C and 30 s at 72°C, then 10 s at 95°C followed by a melt of the product from 65°C to 95°C. The ΔΔCT method was used for statistical analysis to determine gene expression levels. GraphPad Prism 5.0 was used to perform two-tailed Student's t tests.
Western blotting
Protein lysates were corrected for protein content by bicinchoninic acid (BCA) protein assay (Pierce), and western blots were performed for MKS1, ARL13B and INPP5E. β-actin was used as loading control in combination with Coomassie Blue or Ponceau S staining. After dry blotting (iBlot Dry Blotting System, Invitrogen, IB3010-01), the membranes were blocked in 5% powdered skim milk (ELK) in tris buffered saline (TBS) with 0.5% Tween. The primary antibodies (rabbit anti-MKS1, Proteintech 16206-1-AP, 1:3000, rabbit anti-ARL13B, Proteintech 17711-1-AP, 1:1000, rabbit anti-INPP5E, Proteintech 17797-1-AP, 1:1000) and mouse anti-β-actin AC-15, Sigma A5441, 1:15000) were incubated overnight at 4°C. The secondary horseradish peroxidase (HRP)-conjugated antibodies (DAKO, dilution 1:2000) were incubated for 1 h before imaging with enhanced chemiluminescence (ECL) Chemiluminescent Peroxidase Substrate kit (Sigma, CPS1120-1KT) and scanning with a BioRad ChemiDoc XRS+ device with Image Lab software V.4.0, or using film.
IMCD3 spheroid growth assay
After siRNA transfection cells were mixed 1:1 with growth factor-depleted matrigel (BD Bioscience). The IMCD3 spheroids were stained as previously described.42 Primary antibody used: rat anti-ZO1, Santa Cruz sc-3725 (1:500), rabbit anti-β-catenin, BD Bioscience AHO0462 (1:500) and mouse anti-acetylated tubulin, Sigma T6793 (1:20 000) Images were taken with a Zeiss LSM700 confocal microscope and 50 spheroids per condition were scored. Data was normalised to IMCD3 cells transfected with siControl and empty vector, which was set to 1. GraphPad Prism 5.0 was used to perform one-way analysis of variance (ANOVA) with Dunnett's post hoc testing per siRNA treated group of samples.
Immunofluorescence
IMCD3 cells grown on coverslips were fixed for 5 min with ice cold methanol followed by a 1 h blocking step in 1% bovine serum albumin (BSA)/phosphate buffered saline (PBS). Primary antibody incubations (rabbit anti-pericentrin, Novus Biologicals NB 100-68277, at 1:500, rabbit anti-MKS1, Proteintech 16206-1-AP, at 1:300, mouse anti-acetylated tubulin, Sigma T6793, at 1:20 000) were performed overnight at 4°C. Alexa Fluor conjugated secondary antibodies (Life Technologies) were performed for 1 h at room temperture (RT). Coverslips were mounted using Fluoromount G (Cell Lab, Beckman Coulter). Confocal imaging was performed using Zeiss LSM700 Confocal laser microscope and images were processed with the LSM Zen software. Approximately 250 events per condition were scored. GraphPad Prism 5.0 was used to perform two-tailed Student's t tests or one-way ANOVA tests.
Retinal pigment epithelial cells or fibroblasts were grown to 80% confluency and then serum starved for 48 h. Cells were fixed with 4% paraformaldehyde (PFA) for 5 min at room temperature followed by ice cold methanol for 4 min at −20°C. Cells were blocked in PBS containing 10% normal donkey serum (NDS), 1% BSA and 0.1% triton X-100 for 60 min. Fixed cells were incubated in primary antibodies diluted in block (mouse anti-acetylated Tubulin, Sigma T6793, 1:1000, rabbit anti-ARL13B, ProteinTech 17711-1-AP, 1:400, rabbit anti-INPP5E, ProteinTech 17797-1-AP, 1:2000, goat anti-γ tubulin, Santa Cruz sc-7396, 1:200, guinea pig anti-RPGRIP1L, 1:50043) for 80 min at RT and Alexa Fluor conjugated secondary antibodies (Life Technologies) for 45 min at RT. Coverslips were mounted using Fluoromount G with 4',6-diamidino-2-phenylindole (DAPI) (Southern Biotech 0100-20). Fourteen image z-stacks with 0.3 µm spacing were taken with a CoolSNAP HQ2 digital monochrome camera (Photometrics, Tucson,Arizona, USA) through a Marianas live cell imaging system (Intelligent Imaging Innovations, Denver, Colorado, USA) using a Plan Apochromat 63X, 1.4 NA oil objective, using identical capture conditions for mutant and control cell lines. Length and intensity measurements were made in FIJI on 16-bit sum-projection images of z-stacks (see online supplementary figure S1 for flow diagram of methods). Using acetylated tubulin signal to identify the axoneme, we manually painted a mask over each cilium using a 3 pixel-wide brush. We also defined a 30-pixel diameter circular mask adjacent to each cilium to measure background intensity. The mean pixel intensities of the mask objects were measured in each channel using the region of interest (ROI) manager. To calculate cilium-specific signal for each protein (ARL13B and INPP5E), we subtracted the mean intensity in the background ROI from the mean intensity in the cilium ROI. To combine biological replicates, we normalised the cilium-specific average intensities, so that the mean (background-subtracted) intensity for the control line in each experiment equalled 1 (Ctrl-117 or Fetal Ctrl-26153). To measure cilium length, we skeletonised the cilium ROIs and used the maximum branch length function to the longest dimension.
Statistical analysis
One-way ANOVA with Dunnett’s post hoc testing was performed to compare ciliary frequencies of affected fibroblasts and controls. The non-parametric Kruskal-Wallis test (post hoc Dunn’s multiple comparison test) was performed to compare ciliary lengths of affected fibroblast and controls. F-tests were performed to compare variance in ciliary length. CI around medians for non-normally distributed values were obtained by bootstrapping (10000 iterations).
Results
Mutations in MKS1 cause JS
We sequenced all coding exons of MKS1 including at least 2 bps of flanking intronic sequence in a cohort of 435 individuals with JS from 371 families using next-generation targeted sequencing methods.39 Individuals with known causes were not excluded. We identified MKS1 mutations in nine families (table 1 and figure 1). In contrast to previously published mutations identified in fetuses with MKS, most of the mutations in our cohort are not predicted to truncate the protein. All of the single nucleotide changes were identified in <0.02% of a large number of adults without congenital malformations sequenced as part of the NHBLI ESP. Since insertion-deletion variants are not reliably included in the ESP data set, we evaluated 182 neurologically normal controls (Coriell panels NDPT020 and NDPT090—http://ccr.coriell.org), none of whom carried the c.1115_1117delCCT variant. In addition, none of the nine individuals with MKS1 mutations had biallelic rare, deleterious variants in the following genes known to be associated with JS: NPHP1, AHI1, CEP290, RPGRIP1L, TMEM67, ARL13B, CC2D2A, INPP5E, OFD1, TMEM216, TCTN1, TCTN2, KIF7, TMEM237, CEP41, TMEM138, TMEM231, C5ORF42, IFT172, TCTN3, B9D1, C2CD3 and CSPP1.
MKS1 mutations in individuals with Joubert syndrome

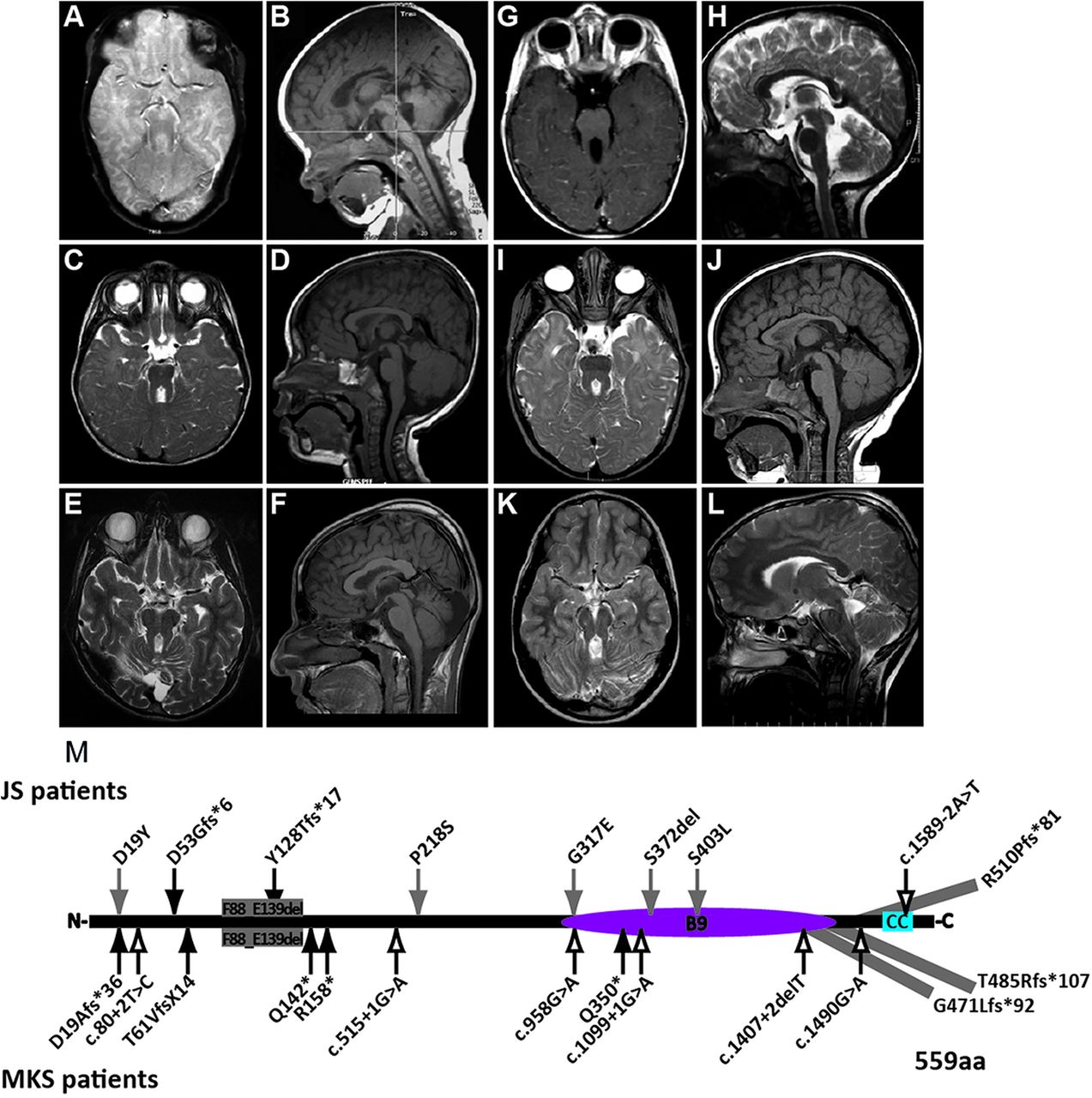
MRI findings and mutations in individuals with MKS1-related Joubert syndrome (JS). (A–L) All affected individuals had classic imaging findings of JS including cerebellar vermis hypoplasia, and thick, horizontally oriented superior cerebellar peduncles. (A and B) is JBTS-10, (C and D) is UW031-3, (E and F) is UW091-3, (G and H) is UW092-3, (I and J) is UW093-3, (K and L) is JBTS-3504 (A, C, E, I and K) are T2-weighted axial views, (G) is a T1-weighted axial view, (B, D, F and J) are T1-weighted sagittal views, (H and L) are T2-weighted sagittal views; (M) MKS1 mutations in individuals with JS and MKS based on sequence NM_017777.3. p.G471Lfs*92 extends the protein by 4 amino acids; p.Thr485Argfs*107 extends the protein by 33 amino acids; p.R510Pfs*81 extends protein by 40 amino acids; MKS1 protein (559 aa). Black arrows, truncating mutations; grey arrows, non-truncating mutations; outline arrows, splice site mutations. CC, coiled-coil; B9, B9-domain.
Individuals with MKS1-related JS rarely have features of MKS
All individuals with MKS1 mutations have characteristic brain imaging findings of JS (figure 1A–L). In addition to the MTS, individuals with JS can have brain abnormalities such as ventriculomegaly, heterotopia, agenesis of the corpus callosum and occipital encephalocele.15 ,44 However, the only other brain imaging abnormality we observed in these individuals was an interpeduncular heterotopia in JBTS-153. Clinically, the affected individuals are indistinguishable from individuals with JS due to other genetic causes but strikingly different from fetuses with MKS (table 1). Only one individual has polydactyly, another has coloboma, and a third has kidney and liver disease, while none has other common features of MKS (encephalocele, cleft palate or skeletal dysplasia). Seven of nine individuals are known to be alive at 4–15 years of age (two are lost to follow-up), which is in contrast to individuals with MKS who usually die in utero or neonatally.
Cilium length in fibroblasts from individuals with MKS1-related JS tends to be longer and more variable than controls
To determine the cellular effects of the MKS1 mutations, we evaluated cilium number and length in primary skin fibroblasts from three of the affected individuals (JBTS-10, JBTS-153 and JBTS-3504), a single fetus with MKS (MKS-158; see online supplementary table S1 Subject: Khaddour ‘07:562), as well as the carrier parents of JBTS-3504 (Parent-3229 and Parent-1753) and healthy, non-carrier controls (Ctrl-10 and Ctrl-117 and fetal Ctrl-26153), using acetylated α-tubulin antibody to mark the ciliary axoneme and γ-tubulin antibody to mark the basal body (figure 2A, see online supplementary figure S2). To determine whether defects were specific to loss of MKS1 function, we also evaluated fibroblast lines from patients with JS with biallelic ARL13B (ARL13B-277)31 and INPP5E (INPP5E-171)41 mutations (See Material and Methods; Cell culture section). Typically, 70–90% of control fibroblasts have cilia after 48 h of serum starvation (figure 2B). In contrast, JBTS-10 fibroblasts were only 52.7% ciliated (p<0.05), and MKS-158 fibroblasts were 24.5% ciliated (p<0.001), while ciliation of fibroblasts from the other patients and the parents of JBTS-3504 was not statistically different from the controls. Cilium length is also consistent in controls, typically measuring ∼3 μm (range 0.2–7.6 µm), based on acetylated α-tubulin and ARL13B antibody staining (figure 2A). The median length (L) was significantly longer in fibroblasts from JBTS-10 and JBTS-3504 (∼4 µm; p<0.001). Additionally, all of the affected JBTS fibroblasts made cilia >8μm, which are longer than the cilia of control fibroblasts, demonstrating higher ciliary length variance (σ2; p<0.001; figure 2C). MKS-158 fibroblasts have more variance in ciliary length compared with fetal Ctrl-26153 as well (p<0.001).

Fibroblasts from individuals with MKS1-related Joubert syndrome display primary cilia defects. (A) Immunostaining of fibroblasts derived from skin biopsies of JBTS-10, JBTS-153, JBTS-3504, MKS-158 and controls. ARL13B (green), γ tubulin (g-tub; white) and cilia (acetylated tubulin, red; scale bar 5 µm). Brightness and contrast were identically adjusted across photos for visualisation purposes; original data is in online supplementary figure S2. (B) Quantification of cilia frequency (mean and SEM) in fibroblasts from controls (Ctrl-10, Ctrl-117, Fetal Ctrl-26153), three individuals with JS (JBTS-10, JBTS-153 and JBTS-3504), the carrier parents of JBTS-3504 (Parent-3229 and Parent-1753), one fetus with MKS1-related MKS (MKS-158), and ARL13B (ARL13B-277) and INPP5E (INPP5E-171) mutants. *indicates p<0.05, ***p<0.001 (One-way ANOVA). (C) Kernel density plots depicting distribution of cilia length (x axis) in fibroblasts obtained from individuals with different MKS1 mutations and controls (y axis). Points and error bars represent medians and 99% CIs, respectively. JBTS-10 and JBTS-3504 have longer (L) cilia than the controls Ctrl-10 and Ctrl-117. ***indicates p<0.001 (Kruskal-Wallis test). Variance (σ2) in ciliary length was different between Fetal Ctrl-26153 and MKS-158, and between Ctrl-10 and Ctrl-117 compared with JBTS-10, JBTS-153, JBTS-3504 and ARL13B-277. ***indicates p<0.001 (F-test). Number of cells scored 100–300 in two batches.
Functional effects of MKS1 mutations on primary cilia
Truncating, presumed null-allele, mutations in MKS1 result in the severe MKS phenotype. To address the functional significance of several of the non-truncating MKS1 variants identified in individuals with JS, we used a three-dimensional (3D) mouse IMCD3 cell culture assay previously used to model ciliopathies.42 We validated the siRNA knockdown of Mks1 by RT-qPCR, western blot and immunofluorescence (see online supplementary figure S3A–D). In IMCD3 cells grown as a monolayer, Mks1 knockdown results in decreased ciliation (p<0.004; see online supplementary figure S3B). Next, IMCD3 cells were transfected with control or Mks1 siRNA in conditions promoting 3D spheroid growth. Immunostaining the spheroids for cilia, tight junctions and adherens junctions revealed that ciliation was reduced >50% upon MKS1 depletion (p<0.0003; figure 3A, B). No gross architectural differences of spheroids were found regarding lumen formation and polarisation.42 Rescue experiments were performed by reconstituting MKS1-depleted IMCD3 cells with wild type (WT) and patient-based mutant expression constructs of human MKS1, which is not targeted by the mouse siRNA against Mks1 (see online supplementary figure S3G). The ciliation defect caused by siMks1 was completely rescued by WT MKS1 or MKS1-p.S403L (p<0.01) and partly rescued by MKS1-p.P218S (p<0.06) (figure 3B). A potential dominant negative effect on ciliary frequency was observed after transfection with MKS1-p.D19Y (p<0.001; figure 3A). Expression of the alleles (in a shorter human isoform of MKS1 construct; full-length was not available) was validated by western blot (figure 3C), and shows different migration from the endogenous full-length mouse MKS1 protein. Despite transfection with equal amounts of DNA, expression differed across the mutant constructs, whereas endogenous MKS1 expression and β-actin levels were equal.
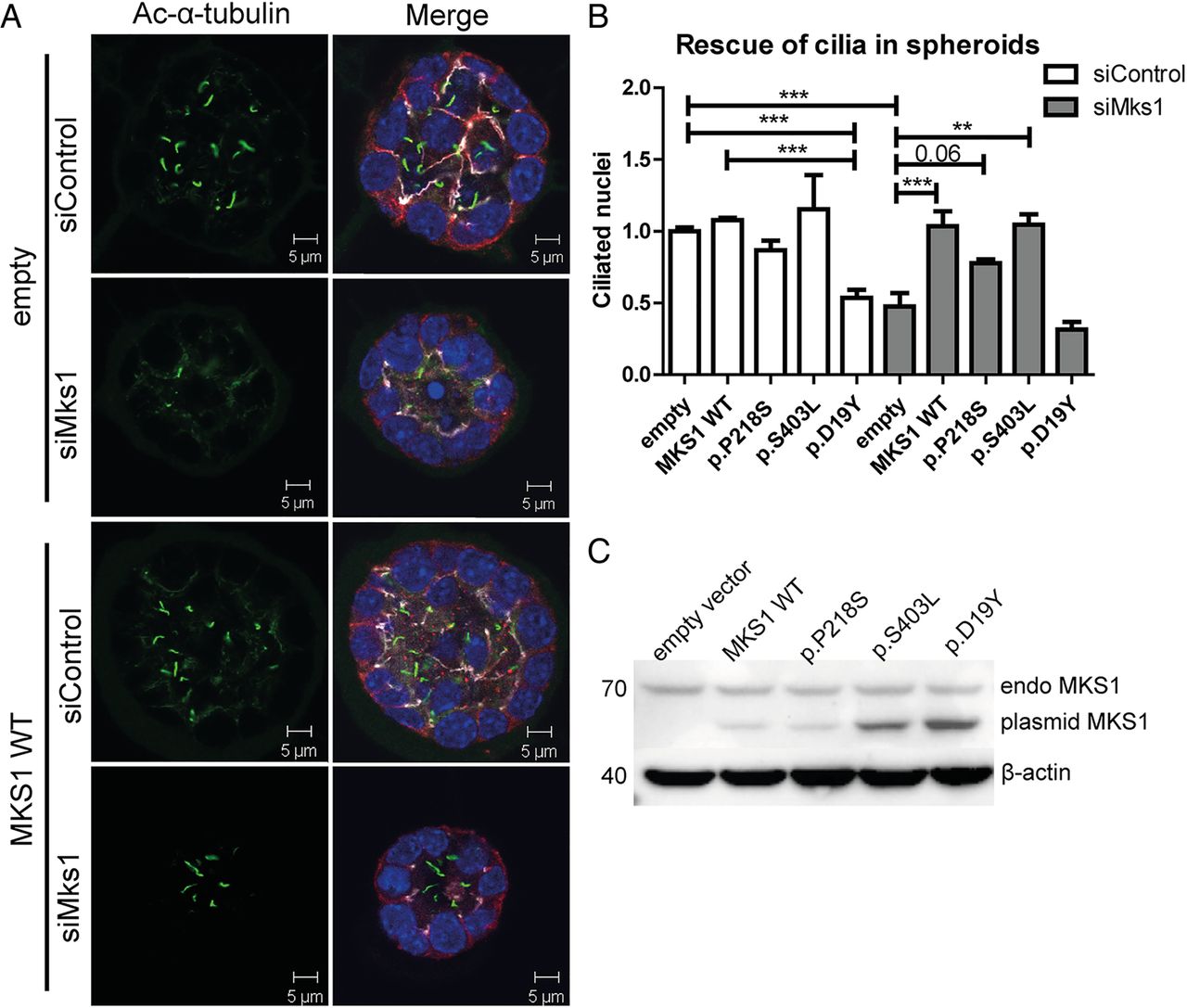
Mks1 knockdown impairs ciliogenesis in three-dimensional spheroid culture of IMCD3 cells. (A) Immunostaining of spheroids for cilia (acetylated tubulin, green), tight junctions (ZO1, white), and adherens junctions (β-catenin, red) with DAPI counterstaining (blue) shows loss of cilia after Mks1 siRNA transfection, and rescue by MKS1-WT. (B) Quantification of ciliary frequency in spheroids shows significant differences between control spheroids and spheroids depleted for MKS1 (indicates p<0.0003), and a potential dominant negative effect of transfection with MKS1-p.D19Y (p<0.001). Complete rescue of ciliary frequency was obtained upon transfection with MKS1-WT or MKS1-p.S403L (p<0.01), and a partial rescue upon transfection with MKS1-p.P218S (p<0.06). Fifty spheroids were scored per condition. Data was normalised to IMCD3 cells transfected with siControl and empty vector, which was set to 1. Error bars represent SEM (n=3 experiments), (C) Immunoblot for MKS1 in IMCD3 lysates (siControl) transfected with different MKS1 alleles. Upper band indicates equal endogenous levels of MKS1 in IMCD3 cells. Lower band indicates different MKS1 alleles (not full-length human MKS1 construct). β-actin is used as loading control. IMCD3, inner medullary collecting duct; WT, wild type.
ARL13B and INPP5E distribution is altered in fibroblasts from individuals with MKS1-related JS
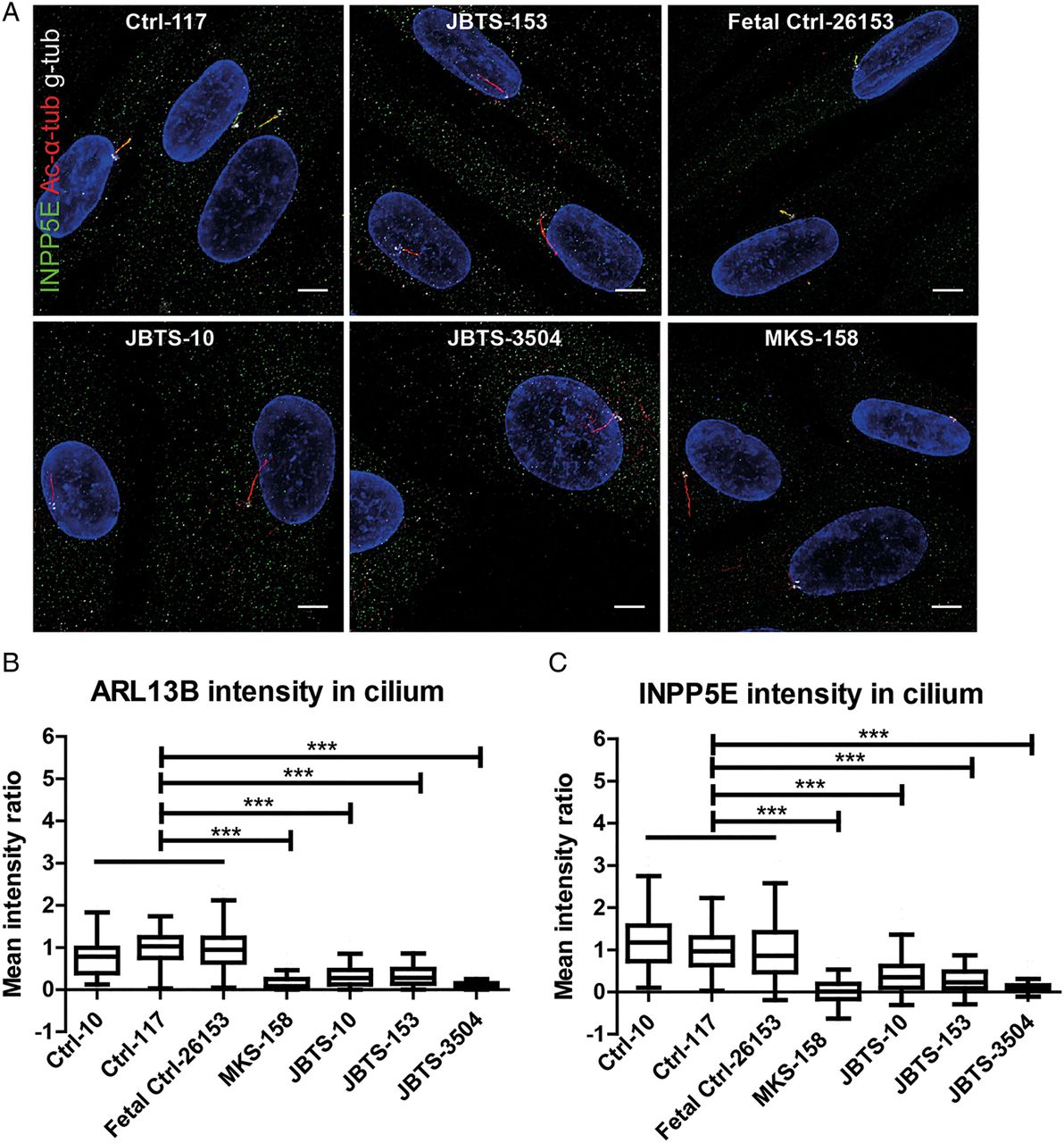
While most of the proteins associated with JS localise to the TZ, where they seem to be involved in the TZ's gatekeeping function, several (ARL13B,31 INPP5E,32 CSPP133 and IFT17234) localise to the cilium. ARL13B is required for ciliary localisation of INPP5E, which extends along the axoneme just distal to the TZ (see online supplementary figure S4E), and which supports the developing hypothesis that mislocalisation of ARL13B and INPP5E is a key part of the mechanism underlying JS.28 ,31 ,32 ,36 To test whether this hypothesis is correct in MKS1-related JS, we evaluated INPP5E localisation in JBTS-10, JBTS-153, JBTS-3504 and MKS-158, and found that INPP5E is markedly reduced in the cilium in all four lines (p<0.001; figure 4A, C and see online supplementary figure S5). Consistent with the known requirement for MKS1 function for ARL13B localisation, ARL13B is also decreased in the cilium (p<0.001; figures 2A and 4B, see online supplementary figure S2). IMCD3 cells transfected with siMks1 show a similar decrease in ciliary ARL13B and INPP5E, supporting the notion that this effect is MKS1 dependent (see online supplementary figure S3E). Of note, decreased ciliary ARL13B and INPP5E are unlikely to be due to decreased expression since the total amounts of ARL13B and INPP5E protein are equal in whole cell lysates from siControl and siMks-transfected IMCD3 cells (see online supplementary figure S3F).

Reduced ciliary ARL13B and INPP5E in fibroblasts from individuals with MKS1-related Joubert syndrome. (A) Immunostaining of fibroblasts derived from skin biopsies of JBTS-10, JBTS-153, JBTS-3504, MKS-158 and controls. INPP5E (green), γ tubulin (g-tub; white) and cilia (acetylated tubulin, red; scale bar 5 µm). Brightness and contrast were identically adjusted across photos for visualisation purposes; original data is in online supplementary figure S5. (B) MKS1-mutant fibroblasts have less ARL13B in the cilium than control cells (Tukey whiskers). ***indicates p<0.001 (Kruskal-Wallis test). (C) MKS1-mutant fibroblasts have less INPP5E in the cilium than control cells (Tukey whiskers). ***indicates p<0.001 (Kruskal-Wallis test). Cilium fluorescence intensity was calculated by subtracting cytoplasmic background from cilium signal and normalising to Ctrl-117 intensity to be able to combine batches (n=50–150 cilia in two batches, see Methods and online supplementary figure S1 for details).
ARL13B-dependent INPP5E localisation is deregulated by MKS1 malfunction at the TZ
Ciliary ARL13B and INPP5E levels were measured in fibroblasts from patients with JS ARL13B-277 and INPP5E-171. ARL13B and INPP5E were decreased in cilia of ARL13B-277 (p<0.001; see online supplementary figures S2, S4A–D and S5). In contrast, only INPP5E was decreased in the cilia of INPP5E-171 (p<0.001; see online supplementary figures S2, S4A–D and S5). These observations in patient fibroblasts, which are consistent with the previously published studies, indicate that INPP5E localisation is downstream of MKS1 and ARL13B function. We next examined MKS1 protein localisation and levels in JBTS-10, JBTS-153, JBTS-3504, INPP5E-171 and MKS-158, and found that MKS1 localises at the TZ of the cilium in all three JBTS lines and controls. On the contrary, levels of MKS1 were decreased at the ciliary TZ of MKS-158 fibroblasts (figure 5A, see online supplementary figure S4F). From this data we conclude that the MKS1 mutations associated with JS do not alter MKS1 protein localisation to the cilium, but the data points to malfunction of MKS1; all residues tested are functionally important as the missense mutations of these residues lead to less ARL13B and INPP5E in the cilium (figure 4). Combined with the fact the INPP5E mutations cause JS, this supports the hypothesis that INPP5E dysfunction may be central in JS (figure 5B).28 ,32 ,36

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MKS1 mutations associated with Joubert syndrome (JS) do not alter MKS1 protein localisation to the ciliary transition zone (TZ). (A) Immunostaining of fibroblasts derived from skin biopsies of JBTS-10, JBTS-153, JBTS-3504, MKS-158, INPP5E-171, and controls for MKS1 (red) and cilia (acetylated tubulin, green). Only the fetus with MKS1-related Meckel-Grüber syndrome (MKS-158) shows decreased MKS1 at the TZ of the cilium. Experiment was performed twice independently. Scale bar 5 µm (B) Schematic overview of the roles of MKS1, ARL13B and INPP5E in JS. Loss of function mutations in MKS1 cause TZ dysfunction and disturbed ciliary localisation of ARL13B and INPP5E. Our data support the hypothesis that loss of ARL13B-dependent localisation of INPP5E is a central mechanism underlying JS. The downstream events are hypothetical based on data in the literature.
Discussion
We report the identification of eight novel MKS1 mutations in nine individuals with JS. Surprisingly, most of the affected individuals show a relatively mild phenotype without features typically associated with MKS. For example, postaxial polydactyly is almost always reported in fetuses with MKS due to MKS1 mutations,45 but it was noted in only one individual with JS (table 1). In addition, we did not observe encephalocele, which is common in MKS1-related MKS. However, one affected individual developed cystic kidney disease and liver fibrosis by 4 years of age, and two developed retinal dystrophy by 2 years and 13 years of age. These findings highlight the importance of monitoring for progressive retinal dystrophy, cystic renal disease and hepatic fibrosis, so that treatment can be initiated before secondary complications occur.
Comparable to other genes that cause JS and MKS (eg, CC2D2A44 ,46 or TMEM6747), individuals with MKS1-related JS carry mutations that are expected to be less damaging than mutations associated with MKS. All nine individuals with MKS1-related JS carry at least one non-truncating mutation (table 1 and figure 1M), in contrast to individuals with MKS1-related MKS, who almost always carry two truncating mutations (see online supplementary table S1 and figure 1M). Indeed, our data support and functionally validate a recent report describing two individuals with mild JS due to MKS1 mutations of which at least one was non-truncating.22 Concordant with the predicted severity of the mutations, the ciliary phenotype is more severe in the fibroblast line from the fetus with MKS compared with the fibroblasts from the three individuals with JS.
Several landmark studies have implicated MKS1 as a component of the B9 protein subcomplex of the TZ at the base of the cilium, which together with other TZ components, is involved in the regulation of protein trafficking in and out of the cilium and sequestering the intraciliary compartment from the cytosol.28 ,29 ,48 Mutations affecting B9 complex proteins have been associated with JS and/or MKS, but not with other ciliopathies, suggesting that this complex has a particular function within the TZ, such as trafficking of INPP5E to the cilium.37 It is likely that mutated MKS1 is partly or entirely degraded in MKS1-associated MKS but, in the case of mild mutations, MKS1 still localizes to the TZ but does not function normal (figure 5A), as was shown for PDE6D mutation as well.37 At the TZ it could cause a disturbance of the lateral diffusion of membrane proteins, resulting in less ARL13B and INPP5E in the cilium. It remains to be investigated how impaired TZ functioning results in more variable ciliary lengths and/or longer cilia.
Complete loss of MKS1 function has been shown to affect cilium formation, likely through effects on basal body docking;49 however, decreased cilium number was not a consistent finding across our affected cell lines. In addition, testing of different alleles in 3D spheroid assays reveals different pathological effects on ciliation. Furthermore, we confirm that ARL13B-mutant and INPP5E-mutant fibroblasts make normal cilia numbers,32 ,36 while CSPP1-mutant fibroblasts make fewer cilia.50 Therefore, decreased cilium number is unlikely to be the primary mechanism underlying JS.
Similarly, altered cilium length is unlikely to be the primary mechanism underlying JS, since we see more variable (and sometimes longer) cilia in MKS1-mutant fibroblasts, while cilia are short in CSPP1-mutant fibroblasts,50 and normal length in INPP5E-mutant fibroblasts.32 More variable cilium length has also been demonstrated in IFT172-mutant21 and ARL13B-mutant (this study) fibroblasts. Given these differences across multiple genetic causes, current data do not support abnormal cilium length as an obligate mechanism underlying JS.
Our data are consistent with the hypothesis that INPP5E dysfunction, either due to mutation or mislocalisation, is an essential part of the mechanism underlying JS (figure 5B).37 This work and previous studies have shown that loss of INPP5E function causes JS.32 INPP5E localisation depends on ARL13B function,36 ,37 and ARL13B localisation depends on TZ function.28 ,29 Recently, loss of INPP5E function in mice has been shown to cause altered phosphatidyl inositol distribution in the cilium and aberrant sonic hedgehog pathway signalling,51 ,52 linking JS-gene dysfunction to sonic hedgehog-related phenotypes such as polydactyly. Although loss of INPP5E function is sufficient to cause JS, mislocalisation of other ciliary proteins due to TZ dysfunction may contribute to the spectrum and severity of phenotypes seen in affected individuals. Aberrant hedgehog and Wnt signalling could contribute to disease development downstream of ciliary dysfunction. Future work will determine the downstream effects of INPP5E dysfunction likely involving inositol phosphate and other signalling pathways, as well as how the many cellular defects associated with loss of JS gene function relate to the human phenotypes. Because INPP5E is potentially druggable, the value of finding this enzyme at the root of JS will hopefully open a novel therapeutic avenue to ameliorate the progression of disease.
Acknowledgments
The authors thank all of the affected individuals, their families and referring physicians for participating in this study. The authors also thank Veronica Foletto and Diana O'Day for technical assistance, and Megan Grout for comments on the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
GGS, CRI and HYK contributed equally. RHG and DD share senior authorship.
Correction notice This article has been corrected since it published Online First. Table 1 subject JBTS-3504 has been corrected.
Contributors GGS, CRI, JCD, GRM, IGP, KJD, JA, IAG, RHG designed and performed experiments. GGS, CRI, HG, SAK analysed data. GGS, HYK, RHG, DD wrote the paper. HYK, DMK, NJM, DN, SDM, JV, LW, NK, SB, MAP, EAO, CAJ, FH, DD provided patient material. NVK, GvH, RHG, DD supervised the work. All authors read and approved the final manuscript.
Funding This research was supported by grants from National Institutes of Health KL2-RR025015, R01NS064077 to DD, the University of Washington Intellectual and Developmental Disabilities Research Center Genetics Core P30HD002274, and DK068306 and RC4-DK090917 to FH and DK090917 to EAO, K23NS45832 to MAP and K24HD046712 to IAG. DD also received private donations from families of children with Joubert syndrome. FH is an investigator of the Howard Hughes Medical Institute. GGS, CAJ, and RHG were supported by grants from the European Union 7th Framework Programme Consortium ‘SYSCILIA’ (241955) and HYK, NVK and RHG receive support from the Dutch Kidney Foundation ‘KOUNCIL’ Consortium Grant CP11.18. HG was supported by the Netherlands Organisation for Scientific Research (ZonMw-TAS grant 116001026), CAJ was also supported by funding from the Sir Jules Thorn Award for Biomedical Research (JTA/09) and a Medical 719 Research Council grant (MR/K011154/1).
Competing interests None declared.
Ethics approval Central committee on research involving human subjects, the Netherlands, CCMO P02.0930C, and the local Ethics Committee of the University of Washington, USA.
Provenance and peer review Not commissioned; externally peer reviewed.