Article Text

Abstract
Background Noonan syndrome is an autosomal dominant, multisystemic disorder caused by dysregulation of the RAS/mitogen activated protein kinase (MAPK) pathway. Heterozygous, pathogenic variants in 11 known genes account for approximately 80% of cases. The identification of novel genes associated with Noonan syndrome has become increasingly challenging, since they might be responsible for very small fractions of the cases.
Methods A cohort of 50 Brazilian probands negative for pathogenic variants in the known genes associated with Noonan syndrome was tested through whole-exome sequencing along with the relatives in the familial cases. Families from the USA and Poland with mutations in the newly identified genes were included subsequently.
Results We identified rare, segregating or de novo missense variants in SOS2 and LZTR1 in 4% and 8%, respectively, of the 50 Brazilian probands. SOS2 and LZTR1 variants were also found to segregate in one American and one Polish family. Notably, SOS2 variants were identified in patients with marked ectodermal involvement, similar to patients with SOS1 mutations.
Conclusions We identified two novel genes, SOS2 and LZTR1, associated with Noonan syndrome, thereby expanding the molecular spectrum of RASopathies. Mutations in these genes are responsible for approximately 3% of all patients with Noonan syndrome. While SOS2 is a natural candidate, because of its homology with SOS1, the functional role of LZTR1 in the RAS/MAPK pathway is not known, and it could not have been identified without the large pedigrees. Additional functional studies are needed to elucidate the role of LZTR1 in RAS/MAPK signalling and in the pathogenesis of Noonan syndrome.
- Noonan Syndrome
- LZTR1
- SOS2
- High-Throughput Nucleotide Sequencing
- Rasopathies
Statistics from Altmetric.com
Introduction
Noonan syndrome (NS (MIM 163950)) is an autosomal dominant disorder characterised by short stature, craniofacial dysmorphism, short and/or webbed neck, cardiac abnormalities, cryptorchidism in men and coagulation defects.1 NS is caused by dysregulation of the RAS/MAPK pathway, which plays a role in diverse biological functions, including proliferation, migration, survival, cell fate determination, differentiation and senescence. Heterozygous pathogenic variants in several genes, PTPN11, KRAS, SOS1, RAF1, SHOC2, NRAS, CBL, BRAF and MAP2K1, account for approximately 75%–80% of all NS cases.2 The clinical phenotype of NS overlaps with other disorders caused by mutations in the RAS/MAPK pathway. These disorders including NS are collectively named RASopathies.3
During the last 2 years, whole-exome sequencing (WES) and whole genome sequencing have been applied to identify disease-causing variants in the remaining 20% of the NS with unknown genetic aetiology. Consequently, pathogenic variants in RIT1 and RASA2 were identified in a small fraction of the cases.4 ,5 These studies underscored the increasing challenges to identify rare, pathogenic variants in disorders comprising high locus heterogeneity, in which the majority of genes have already been identified. In this scenario, to be able to make sense of the large numbers of rare variants typically produced by WES, two reasonable approaches may be applied: one is to study large cohorts and seek candidate genes already known to be involved in the RAS/MAPK pathway, increasing the possibility that more than one individual could harbour rare variants in a common gene; the other is to examine familial cases and search for predicted pathogenic de novo and/or segregating variants without a priori hypotheses regarding the function of the genes.
Herein, these two approaches were applied in a NS Brazilian cohort in order to identify novel genes associated with NS. Subsequently, two additional familial cases from international centres were included.
Methods
Brazilian cohort
Our genetic outpatient clinic follows a large cohort of >200 individuals diagnosed with RASopathies, mainly NS, some of which have been previously reported by our group.6 ,7 They have been screened for mutations in most of the known genes associated with RASopathies by Sanger sequencing or denaturing high performance liquid chromatography, respectively, based on the frequency of the genes in NS and their clinical findings: PTPN11 (ex. 2–15), SOS1 (ex. 1–23), RAF1 (ex 7,14,17), KRAS (ex. 2–6), SHOC2 (ex. 2), CBL (ex. 8–9) and BRAF (ex. 6,11–16). Fifty-eight probands with the clinical diagnosis of NS who fulfilled the diagnostic criteria established by van der Burgt et al8 and who tested negative were selected for WES. Familial cases were prioritised. Written informed consent was obtained prior to collection of samples. Six probands carried mutations in RIT19 and two in NRAS (data not shown). Among the 50 remaining probands, there were four familial cases: a three-generation family with three affected individuals (Br-F3), a mother and five affected children from three different marriages (Br-F4), a deceased mother and two affected siblings (Br-F7) and a daughter–mother pair (Br-F1).
American family
The daughter–mother pair (US-F1) was evaluated for the presence of symptoms suggestive of NS. Molecular testing for all known genes associated with RASopathies using a commercially available next generation panel was negative. Consequently, WES was done in both individuals following written informed consent.
Polish family
A familial case with NS that had no identifiable mutations in the coding exons of PTPN11, SOS1 and RAF1 analysed by Sanger sequencing was selected for WES. They were recruited by an experienced clinical geneticist in outpatient clinic located in Poland and were referred for molecular analysis to the Department of Medical Genetics, Institute of Mother and Child, Warsaw. The study was approved by the local Ethics Committee. The clinical data and DNA samples were obtained with written informed consent for molecular analysis and storage.
Sequencing and filtering of variants
WES of genomic DNA obtained from the peripheral blood of the 58 affected Brazilian individuals and their affected (8) and unaffected (2) available relatives was performed using Illumina's TrueSeq kits for library preparation and exome capture and the Illumina HiSeq sequencer for paired-end reads of approximately 100×100 bp. An average on-target coverage of 60× was achieved. Alignment of the sequences was performed with the Burrows-Wheeler Aligner.10 The Picard and Genome Analysis Tool Kit (GATK)11 were used for data processing and variant calling. Variant annotation was performed with ANNOVAR.12 Filtering was restricted to retain only heterozygous, non-synonymous, exonic and/or splicing variants with a reference population frequency of <0.1%. We used the 1000 Genomes Project National Institutes of Health and the 6500 Exome Sequencing Project Washington University, respectively, as references. In addition, we filtered for an allele frequency of <0.5% among 609 elderly Brazilian controls from our centre (unpublished data). Variants shared by familial cases and variants in genes of the RAS/MAPK pathway were selected for further investigation. Predicted pathogenic variants were Sanger sequenced in available relatives when segregated with the clinical phenotype or to confirm de novo status (figure 1). Paternity and maternity in individuals with de novo mutations (Br-2.1, Br-4.2, Br-5.1 and Br-6.1) were confirmed with polymorphic markers. All variants that were considered pathogenic were confirmed through Sanger sequencing.
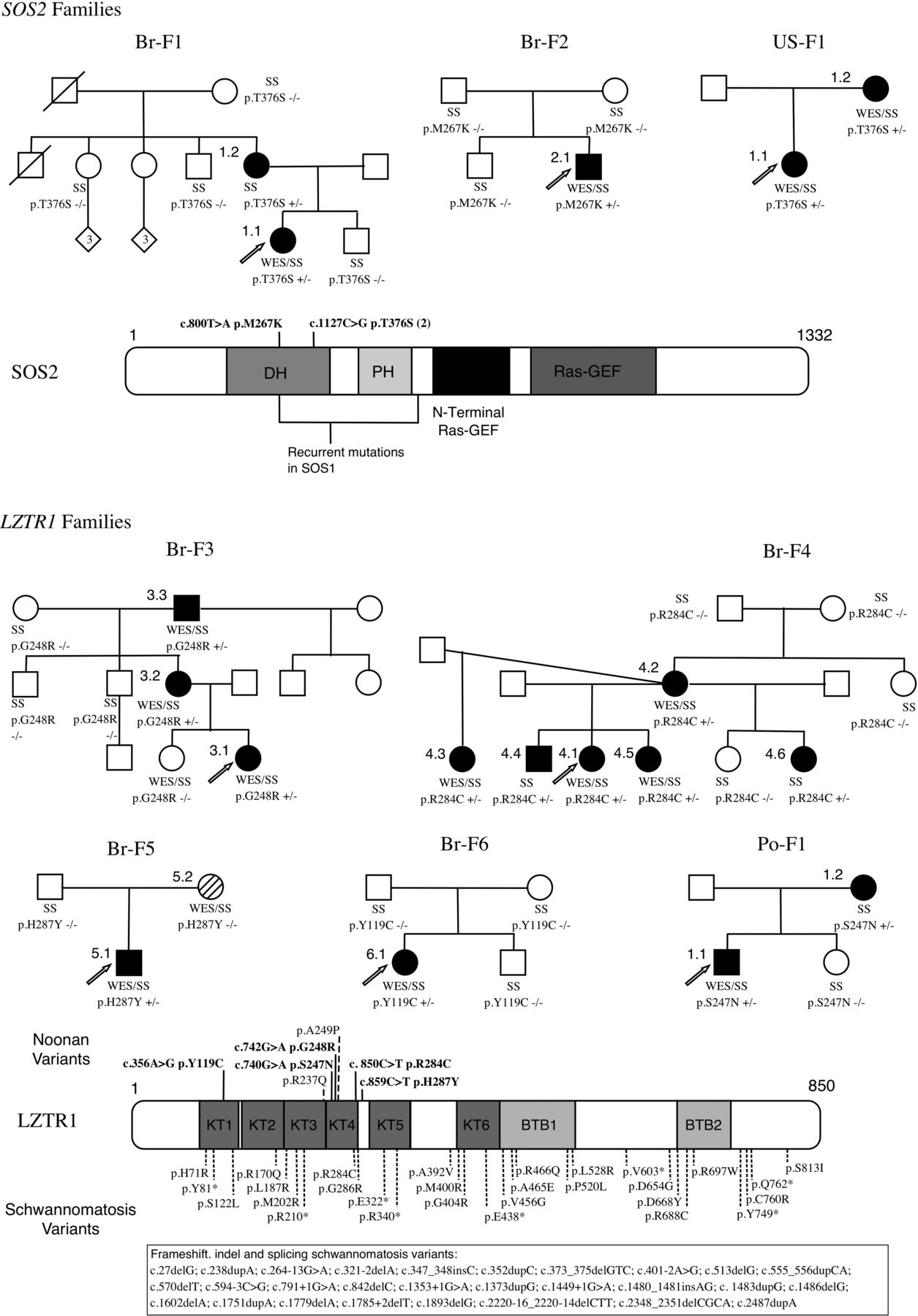
Pedigrees, variants and protein representation of LZTR1 and SOS2. Family pedigrees demonstrating segregation and/or de novo status of the LZTR1 and SOS2 variants. Br-5.2 has neurofibromatosis type 1 (NM_000267:c.2325G>T). Schematic representation of the proteins and their domains showing the mutated residues in the Noonan syndrome and schwannomatosis individuals. In bold: current study, non-bold: previous publications. +/−, heterozygous variant; −/−, homozygous reference; DH, Dbl homology; GEF, guanine nucleotide exchanger factor; PH, pleckstrin homology; SS, Sanger sequencing, WES, whole-exome sequencing.
In silico prediction effects of variants
In silico analysis of variants was performed with open access software, such as SIFT, PolyPhen2, LRT, Mutation Taster and GERP++ algorithms. Variants were considered to be pathogenic by in silico analysis when three or more of these algorithms classified them as damaging.
Statistical methods
Rare variants burden test: two-tailed Fisher's exact test with significance level of p<0.05 was applied to compare frequencies between total number of variants in the 50 proband Brazilian cohort and a WES control database of 107 Brazilian individuals affected by other monogenic disorders.
Results
We performed WES in 50 NS probands without pathogenic variants in genes previously associated with NS. We were able to identify rare, predicted pathogenic variants in two novel genes through the analysis of genes in the RAS/MAPK pathway and the analysis of variants segregating in large families.
SOS2: RAS/MAPK-related gene
Through the analysis of genes belonging to the RAS/MAPK pathway according to the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, we were able to identify SOS2 (MIM 601247; RefSeq accession number NM_006939.2) missense variants in four probands (Br-1.1, Br-2.1, Br-7.1 and Br-8.1) using WES data. Two of them were familial cases (Br-1.1 and Br-7.1). SOS2 is homologous to SOS1, the second gene most frequently associated with NS. All these variants were confirmed by Sanger sequencing, but in family Br-8.1. In silico analysis of the remaining three variants by SIFT, PolyPhen2, LRT, Mutation Taster and GERP++ algorithms predicted pathogenicity in residues that are highly conserved across multiple species (table 1).
Filtered variants in SOS2 and LZTR1
Studies of segregation or de novo status supported pathogenicity in one familial case (Br-F1) and in one isolated case (Br-2.1). In family Br-F7, there were no available relatives to confirm segregation and therefore it was excluded from further analysis. In total, predicted pathogenic variants in SOS2 were identified in two probands in the Brazilian cohort, reflecting a population frequency of 4% (2/50). When comparing the frequency of all identified SOS2 variants (4/50) with the one of controls (3/107), there was no statistically significant difference (p=0.2102). The two heterozygous missense variants, p.M267 K and p.T376S, found in SOS2 in our cohort are both located in the DH domain of the SOS2 protein and p.M267K affects the residue homologous to Met269 in SOS1, a mutation hotspot in NS. The variant p.T376S segregates with the phenotype in Br-F1, whereas p.M267K is de novo in Br-2.1. For comparison, two of the missense heterozygous variants, p.D952N and p.T449A, identified in three controls (all without NS phenotype) are predicted to be either tolerated or benign by in silico analysis. The third variant, p.R334H, is predicted to be probably damaging, with a low frequency in the 6500 exome database. Therefore, even though the burden test was not statistically different from the control population, we consider the variants p.M267K and p.T376S to be pathogenic mutations. Remarkably, the same variant of Br-F1 (p.T376S) was later identified segregating in a family from USA (US-F1; figure 1 and table 1).
LZTR1: a gene not previously associated with the RAS/MAPK pathway
The study of two large families allowed us to identify a candidate gene for NS not formally associated with the RAS/MAPK pathway. In each proband, WES analysis produced approximately 34 000 raw variants in comparison with hg19 reference. After filtering with the aforementioned parameters, each proband still harboured approximately 280 rare, heterozygous missense variants. Including WES data from affected and unaffected relatives of these probands allowed decreasing the number of possibly pathogenic variants. Thus, in family Br-F3, the analysis of the proband, one unaffected and two affected relatives, identified 24 rare variants that segregated with the NS phenotype, while in family Br-F4, with the proband, one unaffected and five affected relatives, only one rare variant was found (see online supplementary table S1). The only gene identified in Br-F4, LZTR1 (MIM 600574; RefSeq accession number NM_006767.3), was also present in the list of rare variants of Br-F3. Analysis of the remaining Brazilian cohort revealed rare variants in LZTR1 in four additional probands (Br-5.1, Br-6.1, Br-7.1 and Br-9.1). The variant in Br-9.1 was considered non-pathogenic, because it was present in unaffected relatives, assuming full penetrance. In Br-F7, the LZTR1 variant had a weak in silico pathogenicity prediction and its segregation analysis was not possible because further relatives were not available. The four remaining missense variants (p.G248R, p.R284C, p.H287Y and p.Y119C) segregated according to the phenotype (Br-F3 and Br-F4) in the familial cases and/or were confirmed to be de novo events (Br-4.2, Br-5.1 and Br-6.1) and in silico analysis predicted pathogenicity. To underscore the association of LZTR1 variants with NS, we performed a rare variant burden test by comparing their frequency in the Brazilian cohort (6/50) versus a control database of WES in 107 Brazilian individuals affected by other monogenic disorders (1/107) and observed a highly significant enrichment (p=0.0044; 2-tailed Fisher's exact test). An additional fifth predicted pathogenic variant (p.S247N) segregating with the phenotype and with damaging in silico prediction was identified in a familial case from Poland (Po-F1; figure 1 and table 1). The five LZTR1 variants (p.G248R, p.R284C, p.H287Y, p.Y119C and p.S247N) are therefore predicted to cause NS.
The clinical findings of the individuals harbouring SOS2 and LZTR1 variants are described in tables 2 and 3, respectively. Their facial photographs are shown in figure 2.
Clinical findings in individuals with SOS2 variants
Clinical findings in individuals with LZTR1 variants

{kind=link}
{kind=link}
Photographs of individuals from different families with Noonan syndrome. Note typical facial features with downslanting palpebral fissures (US-1.1, Br-6.1), hypertelorism (Br-1.1, Br-3.1, Br-4.5) and ptosis (Br-4.1) and short/webbed neck (Br-2.1, Po-1.1). In SOS2 patients there is marked ectodermal involvement (most pronounced in Br-2.1).
Discussion
A large cohort, with additional families from collaborative centres, and large pedigrees provided the basis to identify rare predicted pathogenic variants in two novel NS genes, SOS2 and LZTR1, expanding the currently known molecular spectrum of NS specifically and RASopathies in general.
SOS2
The two known human SOS homologues, SOS1 and SOS2, share 70% homology between them. To date, only mutations in SOS1 were found to be associated with human disorders, including NS and hereditary gingival fibromatosis type 1.13
SOS1, a large multidomain protein, is a RAS-specific guanine nucleotide exchanger factor (GEF) that facilitates the conversion of RAS from the inactive GDP-bound to the active GTP-bound form.13 Pathogenic mutations in SOS1 are the second most common molecular cause of NS. The mainly missense mutations and small in-dels are non-randomly distributed across domains of the protein and were classified by Lepri et al14 in three distinct classes, based on the predicted role of affected residues and functional consequences derived from the nature of the amino acid change. The first class of mutations (class 1A), located in residues that participate in the interaction of the Dbl homology (DH) and Ras Exchange Motif (REM) domains, is predicted to promote conformational rearrangements of these regions that reduce the enzyme self-inhibition by impairing proper masking of the distal RAS binding site or by acting on the allosteric control of catalytic activity. Specifically, the residue Met269, responsible for 10% of total NS cases harbouring SOS1 mutations, interacts directly with residues of the REM domain implicated in RAS binding. The missense heterozygous variants found in SOS2 in our cohort (p.M267K and p.T376S) are located in the DH domain. The residue 267 in SOS2 is homologous to residue 269 in SOS1, providing further evidence to its pathogenic nature. Consequently, we hypothesise that mutations in SOS2 cause NS through a mechanism similar to that of SOS1, that is, gain-of-function mutations leading to enhanced signalling and upregulation of the RAS/MAPK pathway.
There is no strong genotype–phenotype correlation in NS. Nevertheless, individuals harbouring a SOS1 mutation usually present with typical NS craniofacial features and cardiac abnormalities, with pulmonary stenosis and septal defects being the most recurrent findings. However, there is a lower prevalence of short stature and cognitive deficits compared with PTPN11-positive individuals. Ectodermal involvement, including curly hair, sparse eyebrows, hyperkeratosis pilaris and ulerythema ophryogenes, is a hallmark of SOS1 patients among NS.14 ,15 Similarly, our SOS2 cases showed an almost identical clinical phenotype with skin abnormalities, especially ulerythema ophryogenes, which was particularly evident in individual Br-2.1 (figure 2), typical NS facial features, and cardiac defects. In contrast, short stature and learning difficulties were frequent in our cohort. No tumours were observed (table 2). As the number of individuals in our cohort is relatively small, the phenotype in individuals harbouring SOS2 mutations will need to be refined through reports of additional SOS2-positive individuals. From this initial study however, ectodermal involvement seems to be a prominent clinical feature of SOS2 mutations.
LZTR1
LZTR1, leucine-zipper-like transcription regulator 1, encodes a protein member of the BTB-kelch superfamily. Its function is poorly known. It was initially described as a putative transcriptional regulator,16 and later it has been proposed that LZTR1 lacks a BACK domain and colocalises exclusively to the cytoplasmic surface of the Golgi network and not to actin, unlike most other BTB-kelch proteins.17
Our study indicates that rare variants in LZTR1 are responsible for NS. The missense heterozygous variants found in LZTR1 in our cohort (p.G248R, p.R284C, p.H287Y, p.Y119C and p.S247N) are localised in the kelch (KT) domains, especially KT4, and are predicted to be deleterious by in silico analysis. The only variant in LZTR1 identified in the 107 controls (p.P635L) is not within or near the KT domains. Another piece of evidence giving further support to the role of LZTR1 in NS phenotype comes from the study of Chen et al.5 These authors performed WES in 27 NS individuals and two of them harboured rare LZTR1 variants (p.R237Q and p.A249P) in the kelch protein domains (figure 1). However, these variants were not considered responsible for the NS phenotype, since they considered LZTR1 as a gene already associated with a specific disorder, in their case, microdeletion 22q11. We believe that it is likely that these two variants in LZTR1 are responsible for the NS phenotype in both individuals from that study, resulting in a population frequency of 7.4% frequency (2/27), similar to the 8% observed in the Brazilian cohort (4/50).
The association of LZTR1 with human diseases began with the 22q11 microdeletion syndrome, as this gene is localised within the 3 Mb region that is most commonly deleted in the syndrome, but not in the 1.5 Mb deletion that is present in approximately 8% of the cases. The phenotype of these two most frequent deletions is usually indistinguishable. Thus, haploinsufficiency of LZTR1 does not seem critical to the 22q11 phenotype.18
Somatic mutations with loss of heterozygosity in LZTR1 have been associated with glioblastoma multiforme, a malignant central nervous system tumour.19 It has been demonstrated that LZTR1 is an adaptor for CUL3 ubiquitin ligase complexes in a similar manner described for other BTB-KELCH proteins, including KBTBD7 that marks NF1 for degradation and consequently hyperactivates RAS/extracellular signal-regulated kinases (ERK) signalling.20 Moreover, several studies demonstrated that germline loss-of-function variants in LZTR1 predispose to an inherited disorder of multiple schwannomas in a familial cancer model.21–24 LZTR1-related schwannomatosis tumourigenesis requires a germline mutation in LZTR1, a somatic neurofibromatosis type 2 (NF2) variant in cis, and loss of the other 22q allele (or at least a segment containing wild-type LZTR1 and NF2).22 Previously, the same pattern of tumour development has been found in schwannomatosis individuals harbouring germline mutations in SMARCB1. This disorder exhibits a clinical overlap with NF2, but in the latter, bilateral schwannoma of the vestibular nerve is pathognomonic.22 NF1 can also present with neurofibromas, a different type of Schwann cell tumours,25 and both disorders (NF1 and NF2) are caused by loss-of-function mutations in tumour suppressor genes, NF1 and NF2, respectively. Interestingly, KRAS, another gene of the RAS/MAPK pathway besides NF1, has also been implicated as responsible for a case of a NS individual presenting with schwannomatosis.26 Thus, it is possible that the dysregulation of this pathway contributes to the development of schwannomas.
The mechanism by which mutations in LZTR1 confer a NS phenotype is still obscure. Alike tumourigenesis, all genes responsible for RASopathies described thus far cause dysregulation of the RAS/MAPK pathway by increasing ERK signalling, either by gain-of-function mutations in RAS genes and RAS-GEFs, such as PTPN11 and SOS1, or by loss-of-function mutations in GTPase activating proteins, such as NF1.2 Similarly, it is fair to assume that missense heterozygous variants in LZTR1 may also lead to an enhanced signal flow through RAS/MAPK pathway. We can rule out haploinsufficiency as the mechanism of NS phenotype since it is not observed in 22q11 microdeletion syndrome and in the familial cases of schwannomatosis that harbour germline loss-of-function mutations.18 The gene variants in LZTR1 reported in schwannomatosis patients include frameshift (19/62) and nonsense (9/62), together with splicing (9/62) and missense (24/62) mutations scattered throughout the gene.21–24 There is no overlap with the variants found in our NS individuals, with the exception of p.R284C. As NS shows highly variable expressivity, it would be interesting to confirm whether the woman with schwannomatosis harbouring p.R284C reported by Paganini et al22 does not show NS features.
It has been demonstrated that the development of schwannomas requires loss of both functional LZTR1 alleles, which implies that it has a tumour suppressor function. In that sense if somatic mutations leading to complete loss of protein function are required for tumourigenesis, we could hypothesise that to develop a NS phenotype, germline loss of >50% of protein function, in a dominant negative manner, would be required. The fact that the NS individual Po-1.2 from our study developed schwannomas in the right arm gives further support to the hypothesis that his germline variant in LZTR1 is more likely to be a loss-of-function mutation that would lead to the development of tumours when further somatic hits, possibly in NF2 for instance, are added. Unfortunately, material from the schwannoma from Po-1.2 individual was not available, preventing molecular testing. Another fact that corroborates the hypothesis that NS LZTR1 mutations have a negative effect on LZTR1 tumour suppressor function is that two of the variants identified in our cohort were also described in malignant tumour samples: p.G248R (glioma, large intestine carcinoma and melanoma) and p.R284C (endometrium carcinoma) in the catalogue of somatic mutations in cancer (COSMIC database, http://www.sanger.ac.uk/cosmic).
Nevertheless, functional studies are required to unravel the precise role of LZTR1 and whether this gene could be coupled with NF1 and NF2 as a tumour suppressor gene acting in the RAS/MAPK pathway, predisposing to both schwannomatosis and NS.
The clinical findings in our probands harbouring LZTR1 mutations comprise typical facial features (figure 2) and cardiac abnormalities (mainly pulmonary stenosis) in all of them, with low frequency of short stature, ectodermal involvement and cognitive disabilities. It is possible that LZTR1 germline mutations causing NS also pose a higher risk for schwannomas development in this population, since one of our NS individual (Po-1.2) developed multiple schwannomas in the right arm (table 3). It remains to be elucidated whether this predisposition could also include malignant tumours, since somatic LZTR1 mutations have been associated both with solid and hematological tumours (COSMIC database).
Further reports are necessary to delineate the complete phenotype in this group of individuals. The first impression is that the clinical phenotype is similar to PTPN11 positive individuals,9 with the exception of short stature, which was not frequent in our cohort.
In summary, we performed WES in a cohort of NS individuals from different populations, including large familial cases, leading to the identification of two novel genes associated with NS. One of them, LZTR1, is not known to belong to the RAS/MAPK pathway. Mutations in SOS2 and LZTR1 were found in approximately 3% of all NS individuals. Still, 15%–20% of the molecular basis of NS remains unexplained. Copy number variations encompassing the locus of one of the known genes associated with NS have been rarely reported27 and could account for a very small amount of the NS unknown aetiology. Alternatively, it remains to be investigated whether digenic inheritance could also play a role in NS aetiology, in which variants in two or more genes of the RAS/MAPK pathway would be required to overcome a threshold of increased ERK signalling, and consequently manifestation of the NS phenotype. In the latter case, variants that are present in control populations, and which are currently not individually considered as causative, may contribute to the disease when jointly present in a single patient.
Acknowledgments
The authors would like to thank the families for their cooperation.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors GLY, MA, MG, MB, SF, MSN, MZ, OB, JM, MRP-B and DRB performed the sequencing studies and bioinformatics analysis. MG, CH, JP, AA, IC, ACM, OB, AALJ, ACP, CAK and DRB contributed clinical samples and information. GLY, MG, OB, JM, MRP-B and DRB wrote and edited the manuscript.
-
Funding The Brazilian group was financially supported by FAPESP 2011/17299-3, CEPID/FAPESP 98/14254-2 and CNPq 302605/2013-4. The Polish group was financially supported by the National Science Centre grants: UMO-2013/09/B/NZ2/03164 and UMO-2011/01/D/NZ5/01347.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethics Committee of Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo/Institute of Mother and Child Warsaw Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.