Article Text

Abstract
Background Fabry disease results from deficient α-galactosidase A activity and globotriaosylceramide accumulation causing renal insufficiency, strokes, hypertrophic cardiomyopathy and early demise. We assessed the 10-year outcome of recombinant α-galactosidase A therapy.
Methods The outcomes (severe clinical events, renal function, cardiac structure) of 52/58 patients with classic Fabry disease from the phase 3 clinical trial and extension study, and the Fabry Registry were evaluated. Disease progression rates for patients with low renal involvement (LRI, n=32) or high renal involvement (HRI, n=20) at baseline were assessed.
Results 81% of patients (42/52) did not experience any severe clinical event during the treatment interval and 94% (49/52) were alive at the end of the study period. Ten patients reported a total of 16 events. Patients classified as LRI started therapy 13 years younger than HRI (mean 25 years vs 38 years). Mean slopes for estimated glomerular filtration rate for LRI and HRI were −1.89 mL/min/1.73 m2/year and −6.82 mL/min/1.73 m2/year, respectively. Overall, the mean left ventricular posterior wall thickness and interventricular septum thickness remained unchanged and normal. Patients who initiated treatment at age ≥40 years exhibited significant increase in left ventricular posterior wall thickness and interventricular septum thickness. Mean plasma globotriaosylceramide normalised within 6 months.
Conclusions This 10-year study documents the effectiveness of agalsidase beta (1 mg/kg/2 weeks) in patients with Fabry disease. Most patients remained alive and event-free. Patients who initiated treatment at a younger age and with less kidney involvement benefited the most from therapy. Patients who initiated treatment at older ages and/or had advanced renal disease experienced disease progression.
- Genetics
- Metabolic disorders
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Fabry disease (OMIM #301500), an X linked, progressive and life-threatening genetic disease, is caused by deficient α-galactosidase A (α-Gal A) activity.1 ,2 Men with severe mutations in the α-Gal A gene (GLA) and virtually no residual α-Gal A activity accumulate globotriaosylceramide (GL-3; also abbreviated Gb3) and other glycosphingolipids in all cell types, and usually become symptomatic in childhood with pain, gastrointestinal disturbances, angiokeratoma, hypohidrosis, proteinuria and other symptoms.1 ,3 As Fabry disease progresses, patients experience renal function deterioration and hypertrophic cardiomyopathy, and are increasingly at risk of developing complications including end-stage renal failure, stroke, cardiac fibrosis, arrhythmias and premature death.1 ,2 ,4–8 Historical data from the era prior to wide availability of renal replacement therapy and other supportive care interventions for complications, and the advent of enzyme replacement therapy (ERT), has shown that the average age of death for 94 untreated men affected with Fabry disease was 41 years.4 Heterozygous women may have disease manifestations, some as severely as male patients,1 ,8 possibly due to skewed X-chromosome inactivation.1 ,9
Agalsidase beta (Fabrazyme®, Genzyme, a Sanofi company, Cambridge, Massachusetts, USA) is a recombinant form of human α-Gal A. The amino acid sequence of the recombinant form, as well as the nucleotide sequence which encoded it, are identical to the natural form of α-Gal. Agalsidase beta is used for ERT in Fabry disease and is given intravenously every 2 weeks at a dose of 1 mg/kg body weight.10 ,11 Agalsidase alfa (Replagal, Shire HGT, Cambridge, Massachusetts, USA) is also available, outside of the USA, for treatment of Fabry disease (0.2 mg/kg/2 weeks).12
Agalsidase beta at 1 mg/kg/2 weeks cleared microvascular endothelial GL-3 deposits from the kidney, skin and heart in a 20 week multicentre, randomised, placebo-controlled, double-blind phase 3 clinical trial of 58 patients.10 ,13 In the open-label, 54-month extension study, agalsidase beta stabilised renal function in patients with mild-to-moderate renal involvement.11 Baseline proteinuria (>1 g/24 h), >50% glomerulosclerosis and age >40 years at treatment baseline were identified as important factors that limited renal response to therapy.11
This study investigated the long-term outcomes of 52 of 58 patients with classic Fabry disease from the phase 3 clinical trial of agalsidase beta using aggregate data from the trial and extension study (NCT00074971) and the Fabry Registry (NCT00196742). Severe clinical events, renal function and cardiac structure following treatment with agalsidase beta (1 mg/kg/2 weeks) are presented over a 10-year median follow-up period.
Methods
Study design and participants
Data from 52 patients with Fabry disease (50 men, 2 women) who were enrolled in the phase 3 clinical trial at eight primary study sites in four countries (USA, UK, the Netherlands and France)10 were included in this study. All patients had the classic form of Fabry disease as confirmed by α-Gal A activity testing and α-Gal A gene mutation analysis (sequencing of all exons and exon-intron boundaries of the α-Gal A gene, or specific α-Gal A gene mutation analysis if the family mutation was known) in accordance with local standards (see online supplementary table S1). Data were collected during the phase 3 trial,10 the 54-month open-label extension study11 ,14 and in the Fabry Registry (figure 1). Data from six patients from the phase 3 trial were not available because they were not enrolled in the Fabry Registry (figure 1). The median agalsidase beta treatment duration for the 52 patients was 10 years (25th–75th: 7.3–10.3 years). Patient clinical trial data were collected at protocol-specified intervals, whereas Fabry Registry observational data were reported at variable intervals. Preinfusion serum samples for recombinant human α-Gal A IgG antibody testing were collected periodically during the randomised portion of the phase 3 clinical trial and the extension trial. During the Fabry Registry observation period, physicians determined the frequency of sampling. Detailed analytical methods have been published.14 The baseline was the date of first agalsidase beta infusion. Inclusion/exclusion criteria for participation in the phase 3 clinical trial and the extension study have been described elsewhere.10 ,11 ,14
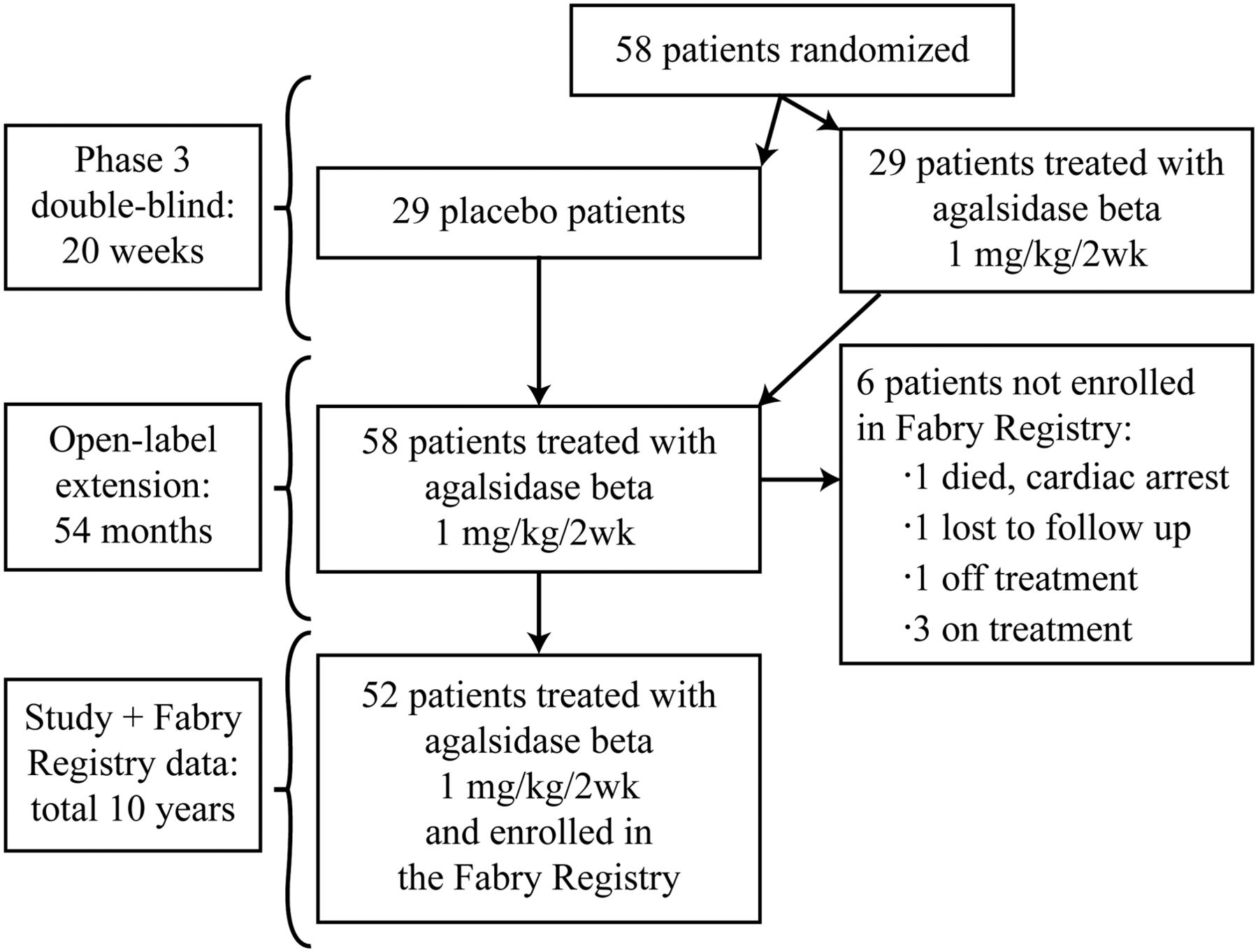
Design of longitudinal follow-up through each of the three study intervals.
Assessments
Severe clinical events were defined as chronic dialysis, kidney transplant, myocardial infarction, congestive heart failure, major cardiac procedures (ie, implantation of a balloon pump, cardioverter-defibrillator or first pacemaker; or bypass surgery), stroke and death. Sites were contacted to ensure that the Fabry Registry contained all available follow-up data.
Estimated glomerular filtration rate (eGFR) was calculated from serum creatinine using the Chronic Kidney Disease Epidemiology Collaboration equation. The urine protein-to-creatinine ratio (UPCR) was used to measure proteinuria. Numbers of glomeruli with global sclerosis or focal segmental glomerulosclerosis were assessed from clinical study biopsies up to the 6 month visit.10
Standard 2-dimensional or M-mode echocardiographic measurements were used to determine left ventricular posterior wall thickness (LPWT) and interventricular septum thickness (IVST).
Statistical analyses
Baseline demographics, other patient characteristics, and severe clinical events were recorded. Estimated GFR, LPWT and IVST slopes and corresponding p values indicating whether slopes were statistically different from zero were calculated using a linear mixed model regression. Patients with a UPCR ≤0.5 g/g and <50% sclerotic glomeruli were classified as low renal involvement (LRI); patients with UPCR >0.5 g/g or ≥50% sclerotic glomeruli at baseline were classified as high renal involvement (HRI). The decision to stratify patients using an UPCR of 0.5 g/g was based on the literature.15 Independent two-sample tests (t test or Wilcoxon) were used to compare HRI versus LRI. Statistical analyses were performed using SAS statistical software V.9.2 (SAS Institute, Cary, North Carolina, USA).
Results
Demographic characteristics of the 52 patients with classic Fabry disease (as confirmed by α-Gal A activity testing and α-Gal A gene mutation analysis) are shown in table 1 (also see online supplementary table S1). The mean age of patients at treatment initiation was 30 years, however those patients classified as having HRI at baseline were over 10 years older than those with LRI (38 years vs 25 years, p<0.001). Eleven of the 52 patients were aged ≥40 years at baseline; eight (one woman) were classified as having HRI and three as LRI (all men). The oldest patient, a man, was 62.2 years of age at baseline and was classified as HRI. Some patients with LRI already had significant evidence of kidney damage at baseline; 13% had ≥20% (but <50%) sclerotic glomeruli.
Demographics and patient characteristics
LPWT at baseline (mean) was higher in patients with HRI compared with patients with LRI (11.9 mm vs 9.8 mm, p=0.005), as was IVST (11.9 mm vs 10 mm, p=0.007) (table 1). While the mean cardiovascular measures of those classified as HRI at baseline were within the normal range, eight patients had abnormal LPWT (>12 mm) and nine abnormal IVST (>12 mm) at baseline; both were abnormal in six patients.
The majority of patients (81%; 42/52) did not experience any severe clinical event during the treatment interval and 94% (49/52) were alive at the end of the 10-year study period. Ten patients (19%) reported 16 severe clinical events, eight during the clinical trial (initial 54 months) and eight during the Fabry Registry observation interval. Severe clinical events that occurred are depicted in figure 2. The most frequent was stroke; five patients (9.6%; four LRI, one HRI) had a total of eight strokes. Four patients (7.7%), all HRI at baseline, had a severe renal event. Two cardiac events were reported, that is, cardiac-related death at age 52 years (patient with LRI), myocardial infarction at age 53 (patient with HRI). Two patients with multiple strokes were in their 20 s at the time of their first severe clinical event. Renal and cardiovascular events occurred most frequently in patients older than 40 years of age. During the initial 5-year clinical trial treatment interval, seven patients had severe clinical events compared with three patients who had their first event in the later follow-up period (figure 2).

Severe clinical events occurring during the study intervals. Renal involvement: LRI=low renal involvement; HRI=high renal involvement. Black symbols: Severe clinical events occurring during the phase 3, randomized, placebo-controlled clinical trial or subsequent open-label extension study. Red symbols: Severe clinical events occurring during the observational Fabry Registry follow-up. Horizontal lines indicate age at first infusion (lower line), and last clinical follow-up visit (upper line).
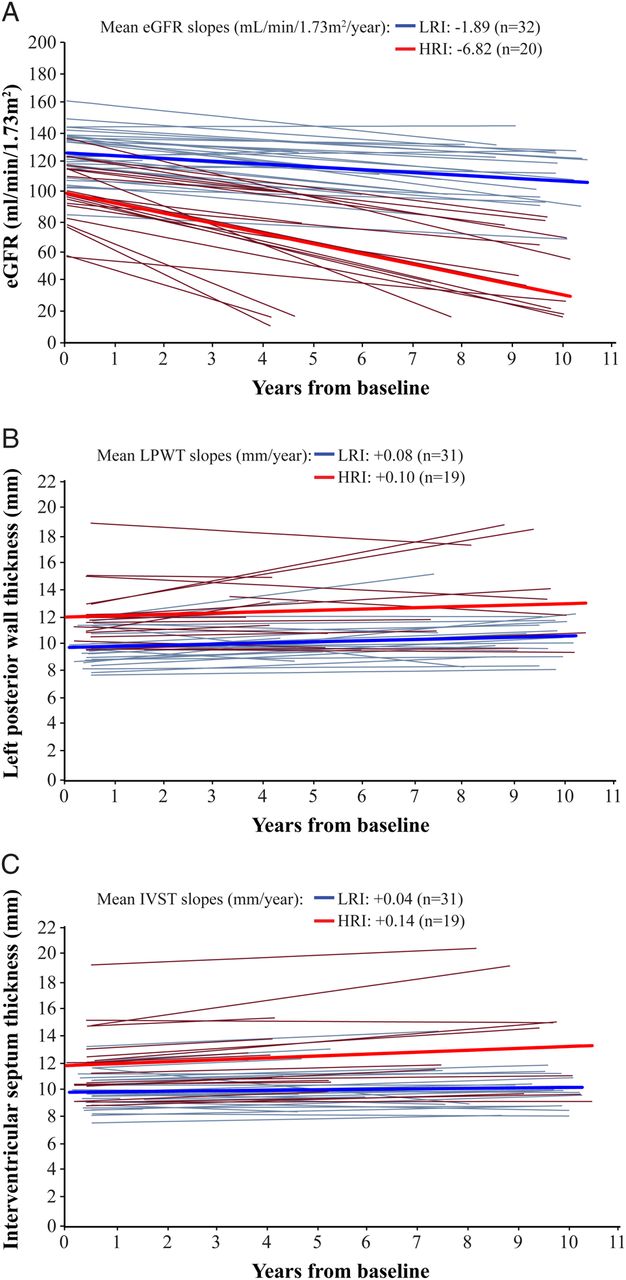
Over the follow-up period, the mean slope for eGFR for the 32 patients in the LRI category was −1.89 mL/min/1.73 m2/year; the mean slope for eGFR for the 20 patients with HRI was −6.82 mL/min/1.73 m2/year (figure 3A). Both were statistically significant (p values 0.001 and <0.001, respectively). Patients with LRI whose UPCR was ≤0.5 g/g throughout the treatment period progressed at a slower rate (−1.48 mL/min/1.73 m2/year, n=20) as compared with patients with LRI whose UPCR rose above 0.5 g/g during the treatment period (−2.6 mL/min/1.73 m2/year, n=12) (p<0.001 for both slopes; data not shown). As reported elsewhere, some of the patients, particularly those with heavy proteinuria or advanced glomerulosclerosis, received ACE inhibitor/angiotensin II receptor blocker therapy at the discretion of the treating physician; however, the effect on proteinuria was variable.11

{kind=link}
{kind=link}
{kind=link}
Estimated glomerular filtration rate (eGFR) slopes (A), left ventricular posterior wall thickness (LPWT) slopes (B), and interventricular septum thickness (IVST) slopes (C). Blue (low renal involvement, LRI) and red (high renal involvement, HRI) bold lines represent the mean slopes of regression lines for the groups. Faint lines represent slopes for individual patients in the respective groups.
Mean LPWT slopes were +0.08 mm/year (p=0.221) and +0.10 mm/year (p=0.282) for patients in the LRI and HRI groups, respectively (figure 3B). Overall, the mean LPWT (n=50) remained within the normal range at the end of the LPWT follow-up (10.6±2.3 mm at baseline, 11.7±3.0 mm at last LPWT follow-up). LPWT did not change over the aggregate treatment interval among patients who were aged <30 years (n=26), or ≥30 years to <40 years (n=13) at the time of first infusion (mean slopes of +0.05 mm/year (p=0.515) and +0.07 mm/year (p=0.494), respectively). Patients who initiated treatment at the age of 40 years or older (n=11) had significant increases in LPWT (mean slope +0.30 mm/year, p=0.013).
Mean IVST slopes were +0.04 mm/year (p=0.536) and +0.14 mm/year (p=0.068) for patients in the LRI and HRI groups, respectively. Both groups showed a similar pattern of minimal change and no statistical significance (figure 3C). Overall, mean IVST (n=50) remained within the normal range at the end of the IVST follow-up (10.7±2.3 mm at baseline, 11.5±3.1 mm at last IVST follow-up). IVST did not change in patients aged <30 years (n=26, slope −0.02 mm/year, p=0.725), increased slightly in patients aged ≥30 years to <40 years (n=13, mean slope +0.17 mm/year, p=0.049), but increased significantly in those aged 40 years or above (n=11, mean slope +0.23 mm/year, p=0.028).
Mean plasma GL-3 normalised within 6 months of treatment and remained normal at last follow-up.
Results of IgG antibody testing during the clinical trial interval have been reported previously.11 ,14 The majority of the male patients seroconverted, most within the first 3 months of receiving agalsidase beta, and showed a downward trend in titres over time, whereas some demonstrated a plateau in their antibody titres or tolerised (no detectable IgG antibodies determined by ELISA and two consecutive negative confirmatory radioimmunoprecipitation assays). In this cohort, seroconversion did not appear to influence the incidence of infusion-associated reactions over time.11 Antibody test results from the Fabry Registry observational period were not available for all patients prohibiting a meaningful analysis.
Discussion
Eighty-one per cent of adult patients with classic Fabry disease receiving agalsidase beta treatment for a median of 10 years remained free of severe clinical events and 94% of patients were alive. This contrasts with severe renal, cardiovascular and cerebrovascular events reported during the fourth and fifth decades of life in ERT-naïve male patients1 ,2 ,4–8 increasing the risk of premature death in their early 50 s.5 ,16 ,17 ERT did not completely prevent the occurrence of severe clinical events. Of the severe clinical events that occurred in the current study, stroke was most frequently reported in patients under 40 years of age. Severe renal events (dialysis, kidney transplantation) were the second most prevalent and primarily occurred in older patients.
We found that patients in the LRI group (mean age 25 years at baseline) experienced some loss of eGFR (−1.89 mL/min/1.73 m2/year) over the 10-year follow-up. Renal disease progression seems to be related, at least in part, to the severity of the disease before treatment. In this study population, some patients with LRI already had significant evidence of kidney damage at baseline, and patients with LRI whose UPCR was ≤0.5 g/g throughout the treatment period progressed at a slower rate as compared with patients with LRI whose UPCR rose above 0.5 g/g during the treatment period. Thus, initiation of ERT before the development of significant glomerulosclerosis and proteinuria may be key to prevent future kidney disease in patients. The reported rate of loss of eGFR in the normal adult population, beginning at age 40 years, is −1 mL/min/1.73 m2/year.18 Untreated male patients with Fabry disease with baseline eGFR >60 mL/min/1.73 m2 were reported to progress at a mean rate of −2.93 mL/min/1.73 m2/year, and severity of proteinuria was found to be associated with more rapid loss of eGFR.2
In the current study, patients with HRI, who began treatment at a mean age of 38 years and had significant pre-existing kidney involvement, showed the greatest decline in eGFR slope (−6.82 mL/min/1.73 m2/year). These data suggest that treatment before major damage to renal architecture occurs is critical and that there may be a point at which impaired glomeruli will inevitably progress to failure. While late-onset treatment may at best slow the progression of renal disease,19 it may still contribute to prolonging life expectancy in advanced, older patients with Fabry disease. In this cohort, severe cardiovascular events were uncommon. This contrasts with previous literature where cardiovascular disease has been reported to be the most common cause of death in untreated patients with Fabry disease.16 ,17
Overall, mean LPWT and IVST did not show a significant increase during the 10-year (median) treatment interval, which suggests stabilisation over time. In patients aged <40 years receiving agalsidase beta, LPWT and IVST remained stable. In contrast, in patients aged >40 years at first infusion, LPWT and IVST significantly progressed from baseline to last follow-up. Although a potential contribution of renal insufficiency or hypertension in the progression of the left ventricular wall thickness cannot be ruled out, these findings are consistent with other research showing that patients initiated on agalsidase beta at a younger age had a favourable cardiac treatment response.20
During the initial 5-year clinical trial treatment interval, seven patients had severe clinical events compared with three patients in the later follow-up period. The current study is the first to report a decline in the number of patients experiencing a severe clinical event after long-term ERT with agalsidase beta. Possible explanations for a decrease in the number of patients experiencing severe clinical events include a long-term benefit from sustained GL-3 clearance21 through agalsidase beta treatment, better adjunctive management of patients, under-reporting of events, or younger age at treatment initiation. As mentioned above, stroke was the most frequently reported severe clinical event in patients under 40 years of age. The effect of ERT on the risk of consecutive strokes remains unknown and recombinant lysosomal enzyme has not been shown to cross the blood-brain barrier.
One study has shown that in patients with advanced Fabry disease, agalsidase beta treatment was associated with a significant 61% relative risk reduction of renal, cardiac and cerebrovascular life-threatening events and death in a per protocol analysis adjusted on proteinuria.19 In comparison, two reports did not show the ability of ERT to greatly influence the outcome in advanced patients, although these studies did not have a randomised placebo group.22 ,23 One report described deterioration of renal, cardiac and cerebrovascular parameters in a cohort of 31 male and 9 female patients treated with agalsidase beta 1 mg/kg for 6 years (median). The mean age of the cohort was 40±9 years. Fifteen events (seven deaths, four cases of end-stage renal disease and four strokes) occurred in 13 patients. Of note, neuropathic pain (n=37), which was not evaluated in our study, improved in 25 patients. The event rate was not different between the ERT group and the untreated (natural history) group retrospectively obtained from the Fabry Registry.23 Median ERT follow-up was shorter than in the present study (6 years vs 10 years, respectively) and included patients that were started on ERT at older ages (mean >38 years).23 In another study of 30 men prospectively followed for a median duration of 5.2 years, renal function declined (−3.4 mL/min/1.73 m2/year, p<0.001) and cardiac mass increased (+1.2 g/m2.7; p<0.001) despite ERT (agalsidase at 0.2 or 1 mg/kg/2 weeks).22 ERT did not prevent the occurrence of cerebral white matter lesions. Comparison of ERT-treated to ERT-untreated patients revealed that for development of a first or second complication the odds declined with longer treatment duration.22 The evidence for effectiveness of ERT in Fabry disease was recently reviewed using a selected set of previously published studies and reported little effect on renal function.24 However, the studies considered in the review did not include baseline renal biopsies or UPCR over follow-up. In addition, there was no stratification by agalsidase alfa and agalsidase beta treatments, and age at which ERT was started. Our results with regard to the ability of agalsidase beta to slow the rate of renal decline in patients with LRI seem to be at variance with the conclusions of this review. These patients followed over 10 years did better than any of the treated groups summarised in the aforementioned review.24 Another study with agalsidase alfa had shorter ERT follow-up (5 years) and did not provide data on severe clinical events.25 It is uncertain if all patients were affected with classic Fabry disease.25
A limitation of this study is that it moved from a randomised, double-blind, controlled clinical trial to a voluntary, observational registry. Data entered into a registry are potentially less reliable than those from thoroughly monitored clinical trials. However, site follow-up occurred on a regular basis during the observational period and physicians were initially and periodically reminded of the importance of timely and accurate reporting.
Also, this study only evaluated severe clinical events, whereas milder clinical events such as non-sustained cardiac arrhythmias, transient ischaemic attacks or hearing loss, which might contribute to disease burden and decreased quality of life of patients, were not analysed. Additionally, over the study period the use of concomitant medications has increased.1 ,26 Adjunctive antiproteinuric (ACE inhibitors/angiotensin II receptor blockers) and antiplatelet therapies, and treatment of comorbidities1 ,27 may have contributed to better outcomes. The current study was not designed to evaluate the impact of concomitant therapies.
This 10-year study documents the long-term effectiveness of agalsidase beta treatment at a dose of 1 mg/kg/14 days in patients with classic Fabry disease. At last follow-up at mean age of 41.6 years, most patients remained event-free and alive. The most favourable treatment responses were observed in younger patients who had less organ damage and started treatment at a younger age. This underlines the importance of early diagnosis and treatment to prevent disease progression and development of irreversible pathological changes. Continued observation will allow further delineation of the long-term perspective of these patients.
Acknowledgments
The authors thank the patients and investigators who participated in the agalsidase beta phase 3 clinical trial and, subsequently, have agreed to participate in the Fabry Registry.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tableS1
Footnotes
-
Contributors DPG, JC, RJD, NG, JK, RHL, GEL, SP, CRS, SW, DGW, NJW and WRW were involved in the initial concept of the manuscript and have reviewed and commented on all drafts and data. DPG, NG, GEL, SW and WRW were involved in phase 3 clinical trial coordination. DPG had full access to the data, wrote the manuscript and had final responsibility for the decision to submit for publication. JK and RL did statistical analysis. Maryam Banikazemi (New York-Presbyterian, University Hospital of Columbia and Cornell, New York, New York, USA) and Karelle Benistan (CHU Raymond Poincaré, Garches, France) collected data and supervised the local research groups. Hans Ebels and Wytske Kingma (Genzyme, Cambridge, Massachusetts, USA) assisted in preparation of the manuscript under the supervision of DPG. Fabry Registry physicians and research coordinators have entered clinical data of these patients included in this study.
-
Funding Genzyme, a Sanofi company, funded the clinical trials and the Fabry Registry.
-
Competing interests Members of the Fabry Registry Board of Advisors (receiving expense reimbursement from Genzyme) include DPG, JC, NG, CRS, SW, DGW and WRW. Authors who have received research funds or travel support from Genzyme include DPG, JC, RJD, NG, RHL, GEL, SW, DGW, NJW and WRW. Authors who have received speaking fees from Genzyme include DPG, JC, RHL, SW, NJW and WRW. DPG has received consulting fees from Shire HGT and Amicus Therapeutics. JC has received consulting fees from Shire HGT and Protalix Corporation. SW has received fees for speaking and for consultancy from Shire HGT and GlaxoSmithKline Pharmaceuticals. RJD is a consultant for Amicus Therapeutics, Genzyme, and Synageva BioPharma, receives grants from Genzyme, receives financial benefit from the sale of agalsidase alfa (Shire HGT) and agalsidase beta (Genzyme), and has founder's stock in Amicus Therapeutics and stock options in Synageva Biopharma. NG has received consulting fees from Genzyme, Shire HGT and Orphan Europe, and is a Member of Registry Boards of Advisors receiving expense reimbursement from Shire HGT and Biomarin Pharmaceuticals. RHL has received educational grants and honoraria from Shire HGT. All honoraria and fees received by GEL are donated to the Gaucher Stichting or the Academic Medical Center Research BV, which both support research in the field of lysosomal storage disorders. SP received educational, programmatic and research support from the Genzyme, Shire HGT, Amicus Therapeutics, Biomarin Pharmaceuticals and Actelion. DGW has served as a paid consultant to Genzyme and also has consultancies and has received travel funds from Abbot, Amgen, Amicus Therapeutics, Gilead, Parion, Relypsa and Shire HGT. NJW receives travel reimbursements and/or honoraria and/or research support from Genzyme, Shire HGT and Pfizer Corporation. WRW has served as a paid consultant to Amicus Therapeutics and Shire HGT. JK and RL are Genzyme employees.
-
Ethics approval The Institutional Review Boards at all sites approved the phase 3 clinical trial and the extension study (NCT00074971), which complied with the Declaration of Helsinki. All patients provided written informed consent before enrolment. Patient participation in the Fabry Registry (NCT00196742) is voluntary. Each independent site was responsible for obtaining informed consent to submit and enter data into the Fabry Registry.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All data are presented in the manuscript.