Article Text

Abstract
Background Whole-genome sequencing (WGS) and whole-exome sequencing (WES) technologies are increasingly used to identify disease-contributing mutations in human genomic studies. It can be a significant challenge to process such data, especially when a large family or cohort is sequenced. Our objective was to develop a big data toolset to efficiently manipulate genome-wide variants, functional annotations and coverage, together with conducting family based sequencing data analysis.
Methods Hadoop is a framework for reliable, scalable, distributed processing of large data sets using MapReduce programming models. Based on Hadoop and HBase, we developed SeqHBase, a big data-based toolset for analysing family based sequencing data to detect de novo, inherited homozygous, or compound heterozygous mutations that may contribute to disease manifestations. SeqHBase takes as input BAM files (for coverage at every site), variant call format (VCF) files (for variant calls) and functional annotations (for variant prioritisation).
Results We applied SeqHBase to a 5-member nuclear family and a 10-member 3-generation family with WGS data, as well as a 4-member nuclear family with WES data. Analysis times were almost linearly scalable with number of data nodes. With 20 data nodes, SeqHBase took about 5 secs to analyse WES familial data and approximately 1 min to analyse WGS familial data.
Conclusions These results demonstrate SeqHBase's high efficiency and scalability, which is necessary as WGS and WES are rapidly becoming standard methods to study the genetics of familial disorders.
- whole-genome sequencing
- whole-exome sequencing
- big data
- de novo mutations
- inherited homozygous or compound heterozygous mutations
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- whole-genome sequencing
- whole-exome sequencing
- big data
- de novo mutations
- inherited homozygous or compound heterozygous mutations
Introduction
Advances in next-generation sequencing (NGS) technologies have made it possible to systematically search for rare disease-contributing genetic variants in human genomic studies. Unlike population-based studies, rare disease-contributing variants can be enriched in families, such as trios (parents and an offspring) or other nuclear families. The availability of family data also facilitates detection of de novo mutations that may be disease-contributing1–4 and provides a valuable resource for NGS studies. Similarly, family based designs can identify many mutations contributing to recessive diseases, inherited as homozygous or compound heterozygous states. Whole-genome sequencing (WGS) and/or whole-exome sequencing (WES) data are being produced at an unprecedented rate, often on families with apparent familial diseases.5 Often, one single study generates multiple terabytes or even petabytes of sequencing data, including raw sequence reads, alignment files, variant calls and annotations. However, the design and development of efficient and scalable computational tools for analysing large sets of sequencing data have lagged far behind our ability to generate data.
Apache Hadoop (http://hadoop.apache.org/) is an open-source infrastructure that allows for reliable and scalable distributed processing of large data sets across clusters of computers using MapReduce programming models. Apache HBase (http://hbase.apache.org/) is an open-source, distributed, versioned and non-relational database modelled after Google's Bigtable.6 Just as Bigtable leverages the distributed data storage provided by the Google File System,7 HBase provides Bigtable-like capabilities on top of Hadoop and Hadoop distributed file systems. HBase is designed to supply random read/write access to structured big data in real time. It can host very large tables, each capable of billions of rows with millions of columns. This capacity can be applied to manipulate hundreds of thousands of WGS samples, which will soon become reality, such as the 100K Genomes Project (http://www.genomicsengland.co.uk/). A number of Hadoop-based tools for processing sequencing data have been developed and applied to NGS studies, such as quality control,8 alignment,9–11 single-nucleotide polymorphism (SNP) calling,10 variant annotation12 and general workflow management.13 In addition, there are many other projects that are built based on Hadoop infrastructure for NGS studies. These include the Hadoop-BAM14 library for manipulating BAM files based on Hadoop, and SeqPig,15 which is a library based on Apache Pig (http://pig.apache.org/) for using the advanced high-level features of Pig to manipulate aligned and unaligned sequence data via writing scripts. However, there is a critical need for a reliable computational toolset that can efficiently manipulate genome-wide sets of variants, their functional annotations and every-site coverage (read depth), as well as analyse NGS data to detect disease-contributing genes based on a scalable big data infrastructure. Leveraging Apache Hadoop and HBase, we have created SeqHBase, a big data-based toolset designed for analysing large family based sequencing data to detect mutations that may be disease-contributing.
Materials and methods
Framework of SeqHBase
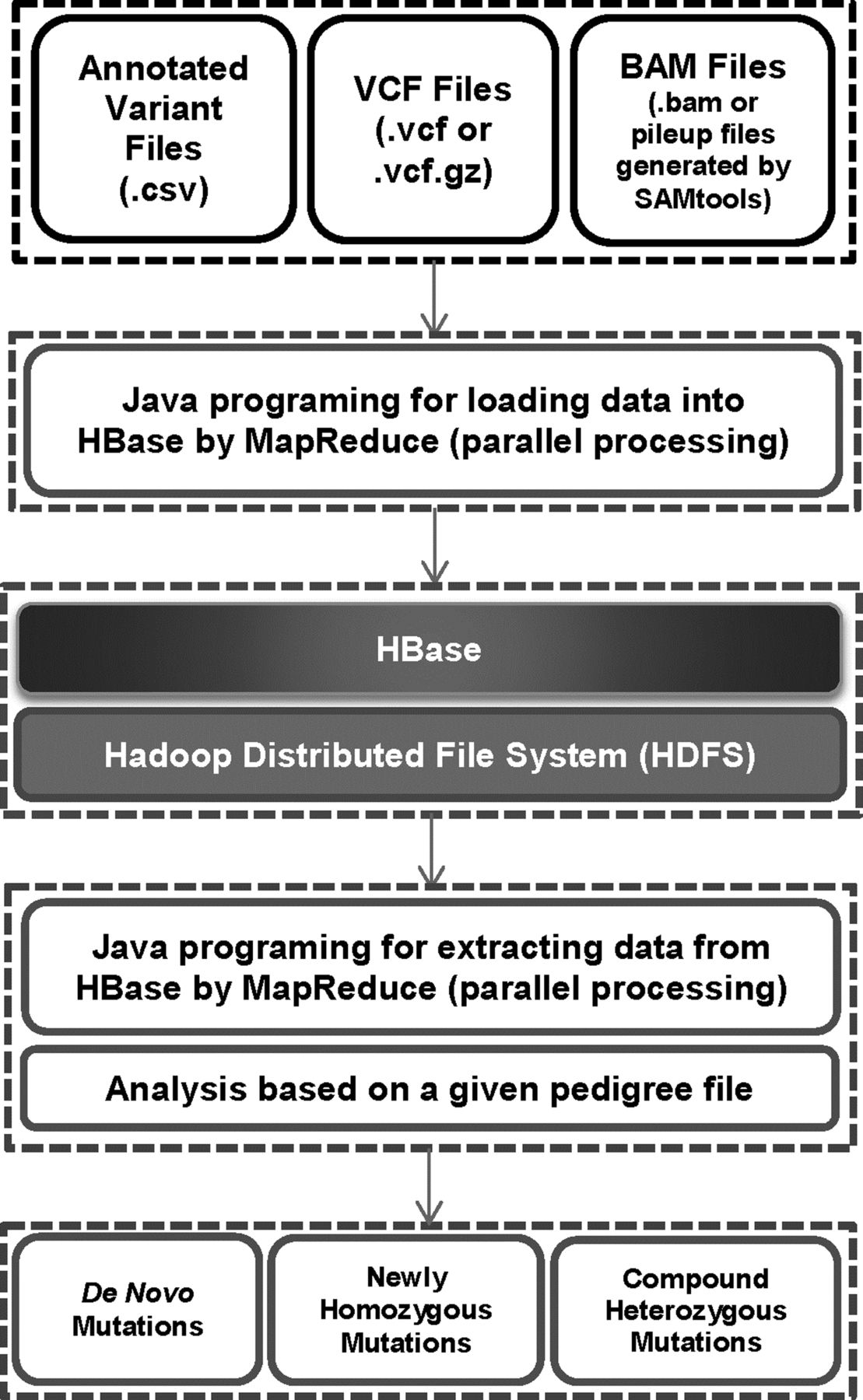
The basic framework of SeqHBase is described in figure 1. SeqHBase manipulates coverage information (every site in the genome) as well as genetic variations and their functional annotations for use in the analysis of each pedigree. Leveraging Apache Hadoop and HBase, SeqHBase efficiently manipulates, stores and retrieves these sequencing data via MapReduce programming models. As described in figure 1, users can load three different types of sequencing data into HBase by providing BAM or pileup files generated by SAMtools16 for coverage information, VCF or vcf.gz files for genetic variations, and comma-separated values (CSV) files for annotated variants. SeqHBase then uses the MapReduce model to split the input data set into independent chunks that are processed by the map tasks in a completely parallel manner. In conjunction with a pedigree file, coverage information, genetic variations and variant annotations are extracted by the reduce tasks in a parallel manner. SeqHBase then generates de novo, inherited homozygous, or compound heterozygous mutations using an analysis engine developed based on the big data infrastructure. SeqHBase was developed in Java and it is freely available for use by academic or non-profit organisations at http://seqhbase.omicspace.org/.

The basic framework of SeqHBase: the sequencing data include annotated variants, genetic variations of every whole-genome sequencing/whole-exome sequencing (WGS/WES) sample, and coverage (read depth) of each site of every WGS/WES sample. Users can load three different types of sequencing data into HBase by providing CSV files for variants, VCF or vcf.gz files for variations, BAM or pileup files generated by SAMtools for coverage. Then SeqHBase uses a MapReduce model to split the input data set into independent chunks that are processed by the map tasks in a completely parallel manner. Given a pedigree file for analysing a data set, SeqHBase extracts variant, variation and coverage information using reduce tasks in a parallel manner for each sample. Finally, SeqHBase uses inheritance information for detecting de novo, inherited homozygous or compound heterozygous mutations that may be disease-contributing in trios, nuclear families and/or extended families.
Extraction of variant, variation and quality information from sequencing data
Variant calling, especially INDELs, along with calling of larger deletions and insertions (>5 bp) is still a considerable challenge.17 In this work, we will not address variant calling. We assume variants are well-called and the VCF and BAM files of every sequencing sample are available. SeqHBase accepts three types of files as input: (1) annotated variant files generated by an annotation programme such as ANNOVAR18 or others. These files can be simple comma or tab-delimited text (.csv) files, where each column represents one piece of annotation information and each row represents one variant. The files are used for extracting variant information, including chromosome number, start position, end position, reference allele, alternative allele, frequency in the 1000 Genome Project19 and/or the EPS6500 project (http://evs.gs.washington.edu/EVS/), ClinVar,20 Combined Annotation Dependent Depletion (CADD) score,21 biological function and multiple diverse function-relevant scores, such as PolyPhen-2 score,22 Sorting Intolerant From Tolerant (SIFT) score23 and others; (2) VCF files for extracting variation information, including sample family ID, individual ID, called variant genotypes, coverage (read depth) and Phred quality scores; (3) BAM files for extracting coverage of each site (∼3 billion sites in a WGS) of every sequencing sample. The diverse types of extracted information are also described in table 1. In downstream analyses, the coverage information can be used to identify if no-call sites are reference-consistent with high quality or reference-inconsistent caused by low quality mapping. A specific function within SeqHBase is developed for directly generating the read depths of each site from BAM files, similar to SAMtools pileup function.
Extracted information from three types of input files
Detection of de novo, inherited homozygous or compound heterozygous mutations
Detection of de novo mutations is based on input parameters provided by users, such as variant frequency in 1000 Genome Project and/or 6500 Exome Sequencing Project (ESP6500) population, minimum read depth, biological functions of interest and predicted functional deleteriousness scores (eg, PolyPhen-2). This filtering step ensures that only functionally relevant variants are examined in the steps below, and the parameters for this filtering step are adjustable by the user. Variants in parents and offspring(s) are examined for all potential de novo mutations where the affected carries a heterozygous variant and both parents carry high coverage (≥20× read depth) reference alleles. Similar to detecting de novo mutations, inherited mutations are also detected based on user-specified input parameters. Parental and affected variations are examined for all potential inherited homozygous mutations, where the affected carries a homozygous variant and both parents carry heterozygous variants. Potential compound heterozygous mutations are examined for every parent-offspring (affected) to ensure that the affected carries two different variants in the same gene region and the contributing variants are each inherited from a different parent. Furthermore, any number of unaffected siblings of the affected can be used to reduce false positive mutations, while any number of affected siblings of the affected can be used to augment the chance of detecting true disease-contributing mutations.
Compilation of three family sequencing data sets
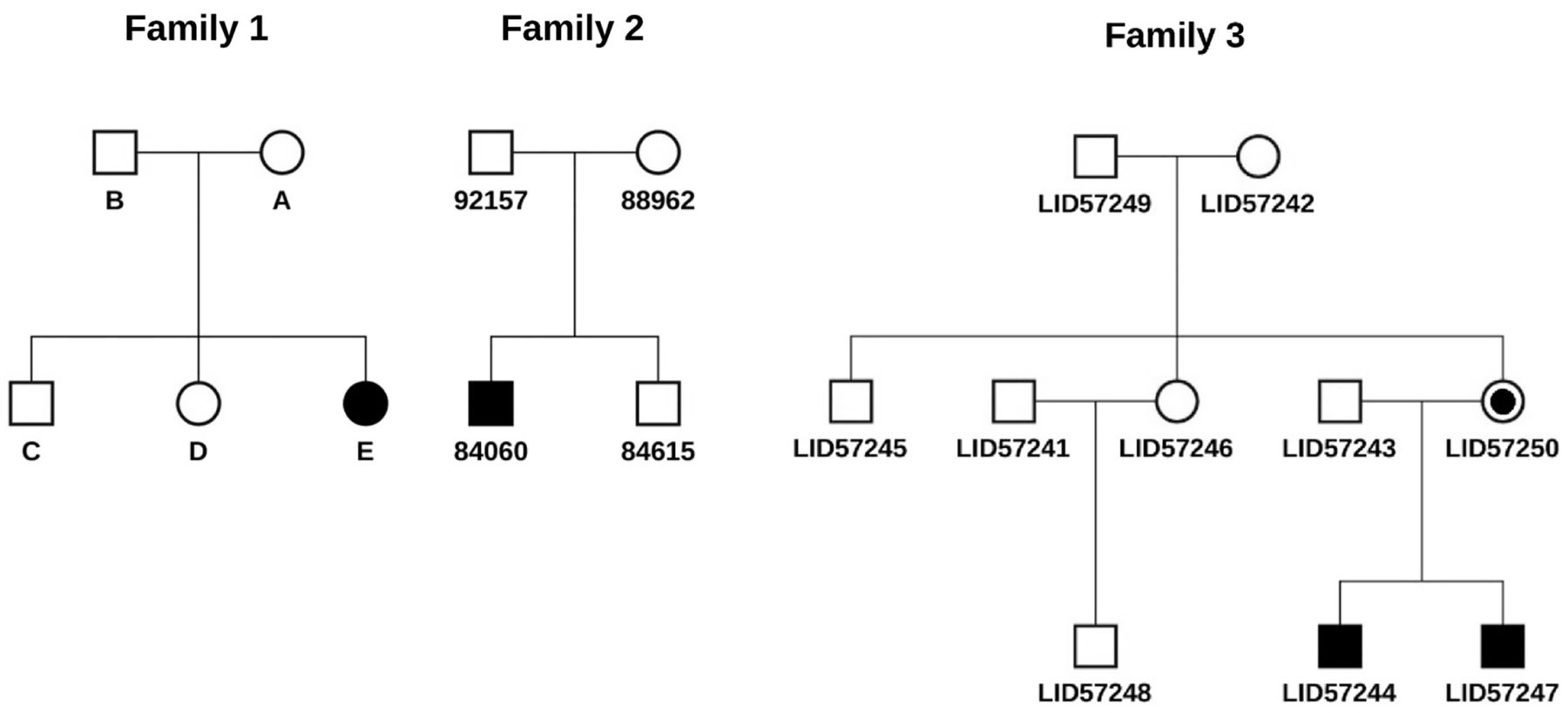
To illustrate the utility of SeqHBase for manipulating sequencing data and detecting de novo, inherited homozygous and compound heterozygous mutations, we analysed three sets of sequencing data from three different family structures described in figure 2. Family 1 in figure 2 is a nuclear family that consists of two unaffected parents, two unaffected children and an affected child (female) with Rodriguez syndrome. The affected in the family had clinical features of severe micrognathia, with a normal chest, short forearms, absent fibula and clubbed feet.24 The samples of the five members in the family were sequenced using next-generation WGS technology by Illumina using a HiSeq sequencer. In addition to providing the BAM and VCF files of the five samples, Illumina also used its quality control pipeline to exclude low quality variants and provided a single nucleotide variant data set that included 3 355 802 clear single nucleotide variants carried by the affected. We then annotated the variants using ANNOVAR. Finally, we loaded the WGS data from the five samples, including the annotated variants carried by the affected, genetic variations of the five samples, and coverage information on each site from the five BAM files, into a Hadoop and HBase cluster built with 20 virtual machines (VMs). We focused on analysing rare variants (eg, variant frequencies ≤0.01 or not observed in the 1000 Genome Project and EPS6500 populations) with interesting biological functions, which included non-synonymous, stop-gain, stop-loss, splicing and frame-shift mutations. The two unaffected siblings’ data were used to systematically exclude false positive mutations by SeqHBase. For instance, if a de novo mutation was present in the affected and either of the two unaffected siblings, the mutation would be excluded from a candidate list in the resulting file, as the mutation is less likely to be a disease-contributing mutation with respect to the syndrome studied.
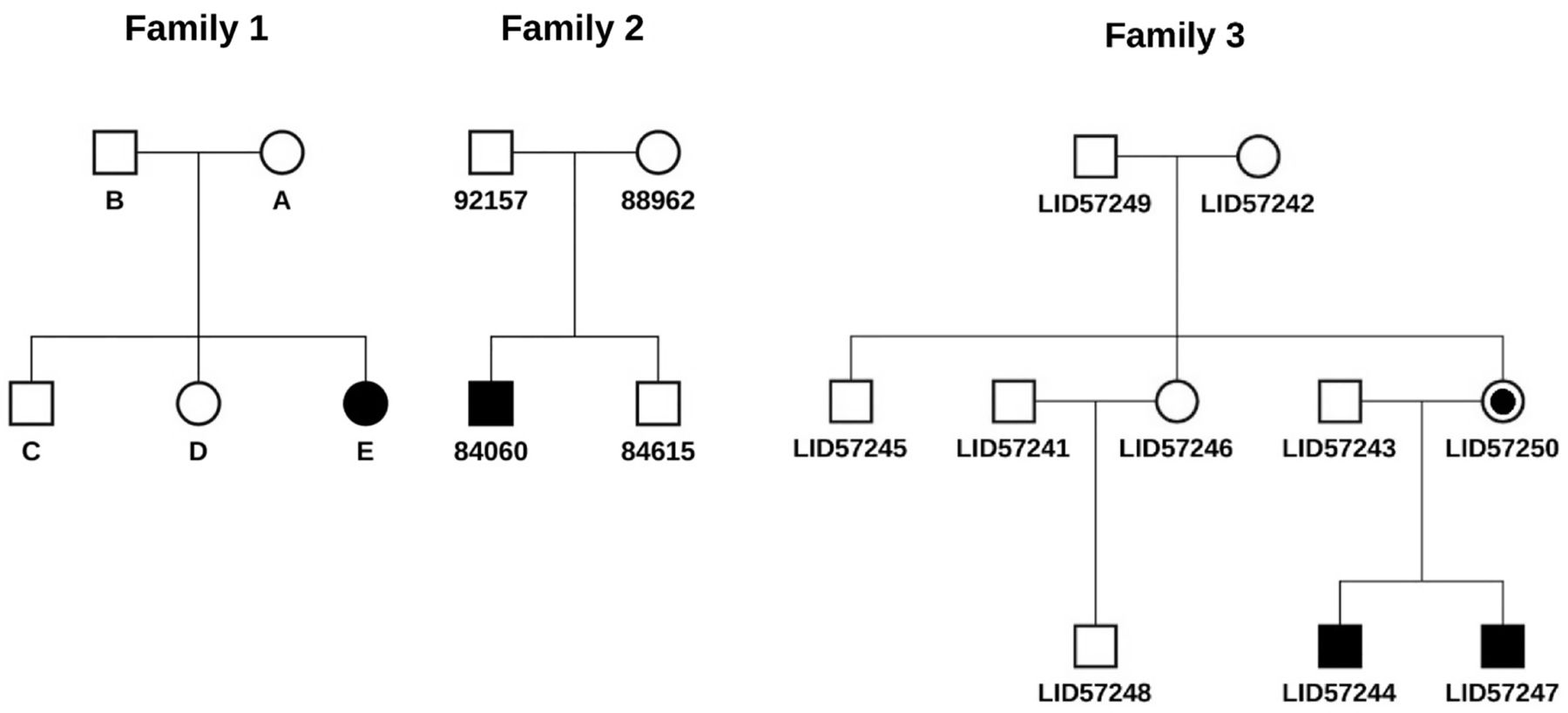
Description of three families used in our benchmarking study. (1) Family 1 is a five-member nuclear family in which the affected individual has Rodriguez syndrome. One plausible de novo mutation and one possible compound heterozygous mutation were detected. (2) Family 2 is a four-member nuclear family where the affected individual has idiopathic haemolytic anaemia. One plausible gene with compound heterozygous mutations was detected. (3) Family 3 is a 10-member extended family with three generations where the two affecteds have an undiagnosed disease manifesting with intellectual disability, autism, attention deficit hyperactivity disorder and other symptoms. An X linked de novo mutation with the mother of the two affecteds was detected. Both affecteds inherited the mutation.
The second data set,25 described in figure 2 as Family 2, was a WES study conducted at the Utah Foundation for Biomedical Research on a four-member nuclear family in which one child (male) was affected with idiopathic haemolytic anaemia (IHA), while his parents and a sibling were unaffected. The annotated variant data, genetic variations and coverage information at every site for the four whole exome samples were loaded into the SeqHBase framework following the procedure described above for the first data set. We analysed rare variants for interesting biological functions in the same manner as the analysis of the first WGS data set. The unaffected sibling's data were used by SeqHBase to systematically exclude false positive mutations.
The third data set (O'Rawe et al, under review), described in figure 2 as Family 3, was a WGS study conducted at the Stanley Institute for Cognitive Genomics at Cold Spring Harbor Laboratory on a 10-member extended family with three generations. The affected individuals are two affected male siblings with severe intellectual disability, autistic behaviours, attention deficit hyperactivity disorder and very distinctive facial features. The other eight members, including grandparents, parents, an uncle, an aunt, an uncle-in-law and a first cousin, are unaffected, without any of the described symptoms. After loading the annotated variant, variation and coverage information into the SeqHBase framework, we analysed rare variants in the same way as described above for the other two data sets.
Results
Performance of SeqHBase
To evaluate the efficiency and performance of SeqHBase, we used 5, 10, 15 and 20 data nodes (VMs) to test the running time of detecting de novo, inherited homozygous or compound heterozygous mutations in the affected individual from the first WGS data set (Family 1 in figure 2), which was composed of a five-member nuclear family. As predicted, the larger the number of data nodes, the shorter the running time with a nearly linear relationship between these two measures. SeqHBase takes approximately 16 s to run analysis on the five-member sequencing data set for detecting de novo and inherited homozygous (or X linked) mutations, and similar time for detecting compound heterozygous mutations. The procedure of detecting de novo and inherited homozygous (or X linked) mutations is separated from the one of detecting compound heterozygous mutations as it was a single variant-based search while the latter was a gene-based search (figure 3).

{kind=link}
{kind=link}
{kind=link}
SeqHBase running time in seconds when run on different numbers of data nodes. The data set for Family 1 in figure 2 was used to evaluate the performance of SeqHBase. Each data node was configured with 6 GB memory, two CPUs (2.6 GHz) and 1 TB hard disk space. Note that the performance of SeqHBase using a single data node is not evaluated due to lack of disk space to manipulate five WGS data sets within the same virtual machine.
Analysis of a WGS data set on a five-member nuclear family
To evaluate the functionality of SeqHBase, we first analysed a WGS data set from a five-member nuclear family that consisted of two unaffected parents, two unaffected children and an affected child with Rodriguez syndrome (Family 1 in figure 2).24 Rodriguez syndrome26 is a rare acrofacial dysostosis syndrome that is clinically distinguished from other acrofacial dysostosis syndromes, such as Miller syndrome27 and Nager syndrome,28 primarily by the severity and distribution of the limb defects.
When first sequenced, a search for de novo mutations within a few interesting gene regions was performed. A de novo mutation in SF3B4 gene was detected and reported.24 We ran a genome-wide search for potential de novo, inherited homozygous or compound heterozygous mutations on the five-sample WGS data set for Family 1 using SeqHBase. After loading the WGS data of the five individuals into a Hadoop and HBase cluster built using 20 VMs, we collected and analysed rare variants with a coverage of ≥20× for every individual, variant frequencies (minor allele frequency)≤0.01 in the 1000 Genome Project and EPS6500 populations, and variants that were annotated as being non-synonymous, stop-gain, stop-loss, splicing or frame-shift changes. Based on the framework built using 20 VMs, SeqHBase took approximately 16 s to scan the whole genome, collect the rare variant list, and generate potential de novo and inherited homozygous (or X linked) mutations. This shows the efficiency and performance of SeqHBase for manipulating and analysing WGS data stored in big tables with multiple billions of records.
When detecting de novo mutations, six candidate mutations were detected. One splicing mutation (chr1:149898811C>T) in SF3B4 (NM_005850:exon4:c.164-1G>A) was the most plausible candidate for an association with Rodriguez syndrome in the pedigree, as expected.29 An inherited homozygous mutation was detected in the analysis using the criteria described above, but the gene (OR56A5) has not shown any association with Nager syndrome or Rodriguez syndrome. When detecting compound heterozygous mutations, two candidates were found. These mutations are briefly summarised in table 2 and shown in more detail in online supplementary table S1. In addition, the two unaffected siblings’ data were used by SeqHBase to systematically exclude false positive mutations. For example, mutations present in both the affected and either of the two unaffected siblings were ruled out as likely disease-contributing mutations. In the procedure of detecting de novo mutations, three false positive de novo mutations were excluded, and in the procedure of detecting compound heterozygous mutations, six false positive compound heterozygous mutations were excluded. This exercise illustrated the clear value of having additional family members in ruling out potential false positive and negative variant calls.
Brief results of family based sequencing data analysis*
Analysis of a WES data set on a four-member nuclear family
A WES data set of a four-member nuclear family was also available to test the functionality of SeqHBase. The nuclear family consisted of two unaffected parents, one unaffected sibling and an affected child who was diagnosed with IHA (Family 2 in figure 2). We previously analysed this data set and identified the disease-contributing gene as PKLR, which carried compound heterozygous disease-contributing mutations in the affected.25 We loaded the WES data of the four individuals into the same Hadoop and HBase cluster built using 20 VMs. We collected and analysed rare variants using the same analysis criteria as was used for analysing the first WGS data set. SeqHBase took approximately 5 s to scan the sequenced regions, collect the rare variant list, and generate potential de novo and inherited homozygous mutations. Similarly, it took approximately 5 s to detect compound heterozygous mutations.
When detecting de novo mutations, 16 candidate mutations were detected. After a thorough literature search for the associated genes, none of them were associated with IHA, as IHA is mainly caused by mutations in genes involved in red cell metabolism (glycolysis, hexose monophosphate shunt and the purine salvage pathway).30 No homozygous (or X linked) mutations were detected in the analysis with the criteria described above. When detecting compound heterozygous mutations, two candidate mutations were detected. The affected carried compound heterozygous mutations, with a rare variant (chr1:155260382C>T) in PKLR (NM_000298:exon11:c.1706G>A:p.R569Q, NM_181871: exon11:c.1613G>A:p.R538Q) inherited from the mother and another rare variant (chr1:155264120C>G) in the same gene (NM_000298:exon7:c.1022G>C:p.G341A, NM_181871:exon7: c.929G>C:p.G310A) inherited from the father. PKLR is a known disease-contributing gene for haemolytic anaemia.31 This compound heterozygous mutation was also reported by Lyon et al,25 and it was replicated by SeqHBase using the analysis criteria described above. Another compound heterozygous mutation was not associated with the syndrome studied. The mutations are briefly summarised in table 2 and shown in more detail in online supplementary table S2. The unaffected sibling's data were used to systematically exclude false positive mutations. In the procedure of detecting de novo mutations, seven false positive de novo mutations were excluded, and in the procedure of detecting compound heterozygous mutations, one false positive compound heterozygous mutation was excluded.
Analysis of a WGS data set on a 10-member three-generation family
To further evaluate the functionality of SeqHBase, we analysed a large WGS data set from an extended family with three generations (Family 3 in figure 2). Two affecteds in the third generation are affected with severe intellectual disability, autistic behaviours, attention deficit hyperactivity disorder and very distinctive facial features. As with analysing the first and second sequencing data sets, the variants carried by the two affecteds were annotated using ANNOVAR and the annotated information was loaded into the same Hadoop and HBase cluster. The genetic variation information was extracted from the VCF files and loaded into the same cluster. The coverage information of every site of the 10 WGS samples was extracted from the BAM files and loaded to the cluster. We ran a genome-wide search for potential de novo, inherited homozygous or compound heterozygous mutations on the 10 WGS data sets using the criteria described in analysing the first data set. SeqHBase took approximately 80 s to collect the rare variant list and generate potential de novo and inherited mutations that might be disease-contributing. And it took similar time to generate a potential compound heterozygous mutation list.
By detecting mutations carried by both of the two affecteds, SeqHBase generated a candidate list for disease-contributing mutations under different genetic models. When detecting de novo mutations, SeqHBase found 18 candidate mutations shared between the two affecteds. When detecting homozygous mutations, one inherited homozygous mutation in the two affecteds was detected in the analysis but the mutations in the gene are not known to be associated with a disease similar to the one studied here. Additionally, one X linked mutation (chrX:70621541T>C) located in TAF1 (NM_001286074:exon25:c.4010T>C:p.I1337T, NM_004606:exon25:c.4010T> C:p.I1337T, NM_138923:exon25:c.3947T>C:p.I1316T) was detected. Interestingly, the X linked non-synonymous mutation in TAF1 was detected as a de novo mutation arising in the mother of the two affecteds. This mutation appears to be a plausible candidate for an association with the syndrome studied in the pedigree, as the gene has been shown to be associated with X linked dystonia-parkinsonism,32 ,33 although further functional study is needed. When detecting compound heterozygous mutations, two candidate mutations, which are carried by the two affecteds individually, located in the same gene were detected. These mutations are briefly summarised in table 2 and shown in more detail in online supplementary table S3. Given the availability of the large pedigree, SeqHBase also used data from other unaffected family members to systematically exclude false positive variant calls. As a result, 14 false positive de novo mutations were excluded as false negatives in the parents of the affected. This example illustrated the power of SeqHBase to generate candidate disease-contributing mutations carried by the two affecteds.
Discussion
Using an Apache Hadoop and HBase framework, we developed a big data toolset for manipulating WGS or WES data. In analysis of three different types of family based sequencing data sets, SeqHBase demonstrated excellent performance with diverse functionality for rapid analysis of familial sequence data.
Although our study mainly focused on family based data sets for candidate disease gene finding using inheritance information, the design of SeqHBase also allows handling of population-based studies with a large number of samples (eg, thousands). Big tables within HBase are capable of handling large-scale information with billions of rows and millions of columns and can be applied to manipulate millions of WGS data sets. Therefore, the design of SeqHBase can be applied to large number of family and/or population-based sequencing studies that are ongoing, such as Epi4K (http://www.epi4k.org/), UK10K (http://www.uk10k.org/) and the 100K Genomes Project (http://www.genomicsengland.co.uk/). The users simply need to ensure that their Hadoop-based framework has an adequate quantity of data nodes for sufficient data storage in the Hadoop distributed file systems. The underlying design of Hadoop MapReduce ensures scalability, flexibility and reliability to handle large data sets as well as analytical applications to process large data sets. Statistical methods for analysing large sets of population-based sequencing data will be developed within SeqHBase in the near future.
ANNOVAR is one of many excellent annotation programmes. Other commonly used annotation programmes include snpEff (http://snpeff.sourceforge.net/), SVA (http://www.svaproject.org/), VEP (http://www.ensembl.org/info/docs/tools/vep/index.html) and others. When developing SeqHBase, we applied ANNOVAR to annotate variants in sequencing data sets. However, annotated information generated by other annotation programmes can also be applied in SeqHBase. Due to the use of open-source framework, SeqHBase is flexible enough to be deployed locally or in the cloud with ease. For example, we have successfully deployed it on Amazon's Cloud (via the Elastic MapReduce service provided by Amazon). For clinical use, sequence data may not be transferred externally to protect patient confidentiality; users can instead build in-house Hadoop clusters in individual labs or at the institutional level and deploy SeqHBase on them.
SeqHBase stores coverage data for all sequenced positions by default, not just for positions with variant calls associated with them. We have chosen this as a design principle, which allows for users to interrogate different sets of variant calls generated for the same sample. Variant calls made on the same sequencing sample are often different based on the variant calling algorithm used, differences in parameter settings used with the same algorithm, use of family aware calling algorithms, or differences in the ensemble of samples used with multisample calling algorithms.17 Therefore, storing coverage data for all sequenced positions allows the users to switch to a different set of variant calls with relative ease. However, we also acknowledge that to save storage space, some users may include only a subset of sites, which can be achieved with user-specified coverage and allelic fraction thresholds.
In summary, SeqHBase is a reliable big data-based computational toolset for efficiently manipulating genome-wide variants, annotations and every-site coverage in NGS studies. It uses a heuristic framework of inheritance information for detecting de novo, inherited homozygous or compound heterozygous mutations that may be disease-contributing in trios, nuclear families and/or extended families. It shows very good performance on three different examples of family based sequencing data and is scalable by virtue of its basis on MapReduce framework. SeqHBase is freely available for use by academic or non-profit organisations at http://seqhbase.omicspace.org/. Source code of SeqHBase can be downloaded after obtaining a license agreement with Marshfield Clinic Applied Sciences.
Acknowledgments
The authors thank Dr Rachel Stankowski and Ms Marie Fleisner in the Office of Scientific Writing and Publication at Marshfield Clinic Research Foundation for assistance with editing and preparation of this manuscript. The authors also thank Mr Ryan Frahm in the Biomedical Informatics Research Center at Marshfield Clinic Research Foundation for building 20 data nodes for this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors Conceived and designed the experiments: MH and KW. Conceived and conducted the first study: EWM, SML and ZY. Conceived and conducted the second study: GJL, RJR and KW. Conceived and conducted the third study: JO, GJL and KW. Performed the experiments: MH, TNP, EH, ZY and JO. Analysed the data: MH, TNP, EH, ZY and JO. Wrote the paper: MH, SJH, EH, SJS, PLP, MHB, JO, GJL and KW.
-
Funding This work was supported by the National Institutes of Health (UL1TR000427 to MHB and MH, HG006465 to KW); the Marshfield Clinic Research Foundation to MH; and the Stanley Institute for Cognitive Genomics at Cold Spring Harbor Laboratory to GJL.
-
Competing interests RJR and KW are share holders and board members of Tute Genomics, Inc. G.J.L serves on the advisory board of Omicia, Inc. and GenePeeks, Inc.
-
Ethics approval Marshfield Clinic Research Foundation; University of Utah; Utah Foundation for Biomedical Research.
-
Provenance and peer review Not commissioned; externally peer reviewed.