Article Text

Abstract
Background Due to high melanoma immunogenicity, germline genetic variants in immune pathways have been studied for association with melanoma prognosis. However, limited candidate selection, inadequate power, or lack of independent validation have hampered the reproducibility of these prior findings, preventing personalised clinical applicability in melanoma prognostication. Our objective was to assess the prognostic utility of genetic variants in immunomodulatory pathways for prediction of melanoma clinical outcomes.
Methods We genotyped 72 tag single nucleotide polymorphisms (SNPs) in 44 immunomodulatory genes in a population sample of 1022 melanoma patients and performed Cox regression analysis to test the association between SNPs and melanoma recurrence-free (RFS) and overall survival (OS). We have further investigated the most significant associations using a fine mapping strategy and followed with functional analyses in CD4+ T cells in a subset of 75 melanoma patients.
Results The most significant associations were found with melanoma OS for rs3024493 in IL10 at chromosome 1q32.1 (heterozygous HR 0.58, 95% CI 0.39 to 0.86; p=0.0006), a variant previously shown to be linked with autoimmune conditions. Multiple additional SNPs at 1q32.1 were also nominally associated with OS confirming at least two independent association signals in this locus. In addition, we found rs3024493 associated with the downregulation of interleukin 10 (IL10) secretion in CD4+ T cells.
Conclusions We discovered novel associations of IL10 with melanoma survival at 1q32.1, suggesting this locus should be considered as a novel melanoma prognostic biomarker with potential for aiding melanoma patient management. Our findings also provide further support for an alternative role of IL10 in stimulation of anti-tumour immune response.
- melanoma
- immune response
- SNP
- survival
Statistics from Altmetric.com
Introduction
The steadily high mortality rate for melanoma1 is in part due to limitations in accurate clinical prognostication of the disease.2 ,3 While current clinical predictors of outcome for early stages of melanoma, such as thickness, ulceration, mitotic rate, and lymph node involvement, have a general applicability,3 there remains large prognostic variability among patients with similar clinical characteristics. This lack of specificity prevents the development of more personalised patient management, an important strategy for potentially reducing melanoma related mortality.
Because of the strongly immunogenic nature of melanoma, in recent years much attention has been centred on exploration of immune surrogates in melanoma progression. Unlike many other solid cancers, T cells mount an antigen-specific response against melanoma,4 manifested by the complex molecular interactions of immune pathways in the tumour microenvironment.5 The expression of pro- and anti-inflammatory cytokines within melanoma and/or in the microenvironment has been shown to correlate with both disease-free (recurrence-free survival, RFS) and overall survival (OS).6 ,7 Similarly, specific profiles of tumour-infiltrating lymphocytes (TILs) have been systematically investigated as potential prognostic markers.8–10 Given this and other evidence, it is highly plausible that the immune response in melanoma patients may be modulated by inherited germline genetic variation thereby impacting disease progression. Previous studies have provided evidence for the association of genetic variation in immunomodulatory genes with patient survival, specifically in interleukins and other pro- and anti-inflammatory cytokines.11–16 While suggestive, these prior efforts were limited by low analytical power and, for the most part, by a lack of independent validation (see online supplementary table S1).
In the current study, we aimed to expand on these prior efforts using a more systematic assessment of the role of germline variants in interleukin and cytokine loci in modulating melanoma clinical outcomes, including attempted validation of previously suggested associations in prior reports. We tested 72 single nucleotide polymorphisms (SNPs) tagging 44 immunomodulatory genes for an association with disease-free (RFS) and overall melanoma survival (OS) in a prospective cohort of 1022 melanoma patients followed by focused genetic and functional analyses of the most significant associations.
Materials and methods
Study population
Biospecimens, clinicopathological information, and demographic information were prospectively collected at the New York University Langone Medical Center under standardised criteria established by the Interdisciplinary Melanoma Cooperative Group (IMCG)17–19 and approved by the institutional review board. All patients provided written, informed consent. Patients diagnosed with stage IV disease were not included in the current study. Patients of self-reported non-European descent were excluded in order to reduce population stratification that could potentially confound the association analyses of the data.
Gene selection, genotyping, and quality control
Candidate immunomodulatory genes were selected from queries of the Gene Ontology database; search terms included ‘interleukins’, ‘regulation of cytokine production involved in inflammatory response’, ‘cytokine secretion involved in immune response’, ‘T cell cytokine production’, and ‘cytokine-mediated signaling pathway’. In total 33 interleukin candidates were selected along with an additional 11 genes with highly relevant roles in cytokine regulation (see online supplementary table S2). To tag these loci, 83 SNPs were selected using Haploview ‘Tagger’20 under an r2 threshold of 0.3 in the HapMap CEU (European ancestry) reference set. For fine mapping of the 1q32.1 locus, an additional 24 SNPs were selected using Haploview with the r2 threshold of 0.6 (see online supplementary table S3). All genotyping was performed by the MassARRAY iPLEX platform (Sequenom Inc, San Diego, California, USA); 1022 samples and 72 SNPs were filtered to a minimum genotyping rate of 90%. Seven SNPs with a minor allele frequency <5% and five SNPs that significantly departed from Hardy-Weinberg equilibrium (p<0.001) were excluded from the analysis.
Statistical analysis
Statistical analyses including univariate and multivariate Cox proportional hazards models and Kaplan-Meier survival estimates were run using the R ‘survival’ package. First, univariate Cox proportional hazards models were used to estimate the HRs for non-genetic variables such as clinicopathological/demographic characteristics, and p values were generated using a likelihood ratio test. The non-genetic variables with a significant effect (p<0.05) in the univariate analysis were incorporated into multivariate Cox models. These included tumour stage, primary tumour thickness, ulceration, histological subtype, age at diagnosis, and gender (see online supplementary table S4). The time from primary diagnosis to first recurrence or last follow-up was used for testing associations with RFS, and time from primary diagnosis to death or last follow-up was used for testing associations with OS. To assess the prognostic potential of the SNPs, each variant was added separately to the model as a genotypic variable with two degrees of freedom; the log-likelihood was calculated to determine a p value. HRs were calculated for heterozygotes and minor allele homozygotes with common homozygotes as a reference. Exploratory analyses for certain SNPs of interest have also been performed using a dominant model, comparing major allele homozygotes against heterozygotes grouped with minor allele homozygotes. For adjustment of multiple testing, a conservative threshold using Bonferroni correction has been used based on the total of 72 SNP comparisons (72 tests; p=0.05/72=0.0007).
Cytokine secretion in CD4+ cells and correlation with rs3024493 genotypes
The secretion of interleukin 10 (IL10), IL2, IL4, IL6, IL17a, interferon (IFN) and tumour necrosis factor (TNF) was assessed in cytotoxic CD4+ cells as per the manufacturer’s protocols. Peripheral blood mononuclear cells (PMBC) were isolated from peripheral blood buffy coats of patients. Blood was layered on top of Ficoll-Paque PLUS (GE Healthcare) and centrifuged at 400×g for 30 min. The mononuclear cell layer was collected, and red blood cells were lysed by the addition of 2 mL ACK buffer (Life Technologies) for 2 min. Isolated PMBCs were resuspended in complete RPMI (Roswell Park Memorial Institute) media plus 20% fetal bovine serum (FBS) and 10% dimethyl sulfoxide (DMSO) and frozen until use. Following thawing, PBMCs were cultured at 5×106 cells/mL in complete RPMI plus 30 U/mL recombinant human IL2 (R&D Systems) for 12–18 h at 37°C. CD4+ T cells were isolated using the EasySep Human CD4+ T cell Enrichment Kit (Stem Cell Technologies), following the manufacturer's instructions. Purified CD4+ cells were then resuspended at 106 cells/mL incomplete RPMI and plated in flat-bottom 96-well plates at 105 cells/well and activated overnight by addition of 2 µL of CD3/CD28 human T cell activation beads (Life Technologies). Supernatants were collected and frozen at −80°C until cytokine analysis. Supernatant IL2, IL4, IL6, IL10, IL17a, IFN, and TNF were quantified by cytokine bead array (BD Biosciences), following the manufacturer's instructions. Beads were analysed by collection on the FACSCalibur cell analyser (BD Biosciences) and quantification using FlowJo software (TreeStar). Spearman's correlation was used to measure the statistical dependence between the average cytokine concentrations (pg/mL) (for each of the seven measured cytokines) and the rs3024493 genotype under both an additive model and a dominant model as described in the Statistical analysis section.
Results
Patient characteristics
The study included 1022 cutaneous melanoma (CM) patients, prospectively accrued by the IMCG at the New York University Langone Medical Center (NYULMC).17–19 Patients’ age at diagnosis ranged from 17–100 with a median age of 58 years and a mean±SD age of 57.7±16.6 years. All cases were of self-reported European descent. Approximately 17% of patients reported a family history of melanoma (n=167), with a higher ratio of males (n=588, 57.4%) compared to females (n=436, 42.6%), similar to the national trend in melanoma incidence rates.2 The median follow-up time was 40.0 months with a total of 162 deaths (18.8%). The majority of primary diagnoses were of stage I (n=685, 66.9%), followed by stage II (n=165, 16.1%), and stage III (n=174, 17.0%). There were no patients with primary diagnosis of stage IV melanoma included in this study. The staging was based on the 2009 American Joint Committee on Cancer (AJCC) TNM classification criteria.3 Additional information was collected on the primary tumour thickness, ulceration, mitotic rate, histological subtype, and anatomic site, as well as the involvement of regional lymph node metastases, and the time for first recurrence. Besides OS, the data on RFS has been collected as an additional clinical outcome; a total of 237 (24.0%) patients had recurrences.
Association of germline variants in immunomodulatory genes with melanoma recurrence-free survival (RFS) and OS
We first examined the prognostic potential of 72 tag SNPs in 44 candidate immunomodulatory genes in predicting RFS among 1022 melanoma patients. Utilising a previously established multivariate Cox proportional hazards model,18 we tested each SNP for correlation with RFS including as covariates: stage, thickness, ulceration, histological subtype, age at diagnosis, and gender. Each SNP was tested separately using a two degrees of freedom test (common homozygotes vs heterozygotes vs rare homozygotes). The strongest association with RFS was observed in multivariate analysis for rs2069840 in IL6 (p=0.0022). Additional associated loci included IL36B, IL21, IL17D, IL25, and IRF8 (table 1). Two tagging SNPs from the IL25 region, rs7145531 and rs3811178, were associated with time-to-recurrence (p=0.0035 and p=0.028, respectively). However, none of the associations with RFS reached the Bonferroni adjusted level of significance (0.05/72, p=0.0007), nor have they validated previously reported findings.
Multivariate association analysis of immunomodulatory germline variants with melanoma recurrence-free survival
We further tested 72 tagging SNPs for their association with melanoma OS. The SNPs in IL10, IL20, IL15, IL13, IL25, IL16, and STAT3 showed associations with OS (table 2). Interestingly, the top two strongest associations were observed for SNPs tagging the interleukin locus at chromosome 1q32.1: rs3024493 (p=0.0006) and rs2222202 (p=0.0042). For rs3024493, the association surpasses the Bonferroni adjusted level of significance. However, it does not show an additive trend as the effect is in the opposite direction in heterozygotes (HR 0.58, 95% CI 0.39 to 0.86) compared to minor allele homozygotes (HR 4.73, 95% CI 1.68 to 13.29). As a result, the significance of the association for this SNP under a dominant model (combining heterozygotes with minor allele homozygotes versus major allele homozygotes) dropped to a nominal level (HR 0.64, 95% CI 0.44 to 0.94; p=0.02) (see online supplementary figure S1 for Kaplan-Meier plots). In contrast, the second most significant association with melanoma OS, rs2222202, does show an additive effect in the protective direction (heterozygote HR 0.58, 95% CI 0.40 to 0.84; minor allele homozygote HR=0.50, 95% CI 0.31 to 0.81), suggesting that the minor allele of rs2222202 associates with more increased survival. Less than 100 kbp away, another SNP from this locus (1q32.1) tagging IL20 was also associated with OS at a nominal level of significance (rs1400986, p=0.041). This region at 1q32.1 contains four interleukin genes spanning ∼150 kbp (IL10, IL19, IL20, and IL24). While the linkage disequilibrium (LD) between each of these SNPs is weak (r2<0.3), we tested whether rs2222202 and rs1400986 were independent of rs3024493. After conditioning the analyses on the status of rs3024493 genotype we found that both SNP associations remained nominally significant, although the strength of the signal for rs2222202 was reduced (rs2222202, p=0.035; rs1400986, p=0.016), indicating that both SNPs may represent the same association signal. Hence, our study provides evidence for a novel association with melanoma OS for rs3024493 in 1q32.1.
The multivariate association analysis of immunomodulatory germline variants with melanoma overall survival
Fine mapping of the most significant association in interleukin locus at 1q32.1
Given that the most significant associations in our study were observed for multiple SNPs at 1q32.1, we genotyped additional SNP tags to refine the associations and to test the relationship between the multiple association signals observed for this locus. Twenty-four SNPs at an r2 threshold of <0.6 were selected to capture the common (minor allele frequency (MAF) >5%) variation in the association region of ∼175 kbp at 1q32.1 containing IL10, IL19, IL20, and IL24. The SNPs were genotyped in the same cohort of 1022 patients. Five additional SNPs from this locus were associated with OS using a genotype test (rs1800890, p=0.00082; rs960326, p=0.0032; rs1800896, p=0.0067; rs3024491, p=0.0086; rs1028182, p=0.022) (table 3, figure 1). Four of the SNPs in the region (here referred to as rs1800890 block) show a comparable effect size and are in strong LD (r2>0.7, table 3): rs2222202, rs3024491, rs1800896, rs1800890. The most significant effect from this block was observed for rs1800890 (rare homozygote HR 0.46, 95% CI 0.24 to 0.88; p=0.00082, see online supplementary figure S2), on the borderline of Bonferroni adjusted p value threshold for multiple testing. After fine mapping analysis, rs3024493 still remains the strongest association in the region (table 3, figure 1A), and shows only a weak LD with the rs1800890 block (r2<0.29). The rs3024493 and variants from the rs1800890 block map within a narrow region (<6 kbp) surrounding IL10 (figure 1). Upon conditioning the models on the genotype of rs3024493, two SNPs in the rs1800890 block (rs3024491 and rs1800896) were no longer significant, while rs2222202 and rs1800890 remain in the nominal range (rs2222202, p=0.03; rs1800890, p=0.03) (table 4, figure 1B). This indicates that despite the weak LD between rs3024493 and the rs1800890 block, these five SNPs in IL10 may represent the same association signal. Besides the SNPs in IL10, there was another association signal in this locus in the fine mapping analysis observed near IL19 (64 kbp away from IL10) driven by three SNPs (here referred to as rs960326 block): rs1028182, rs960326 and rs1400986, with the strongest effect observed for rs960326 (minor allele homozygotes HR 2.90, 95% CI 0.67 to 12.57; p=0.003) (table 3, figure 1A). While there is a weak LD between rs1028182 and rs960326 (r2<0.29), rs1400986 is not correlated with the other two SNPs (r2<0.01). Similarly, none of the IL10 SNPs correlate with these three variants (r2<0.01). In addition, when conditioned by the status of rs3024493, the significance for the three SNPs in rs960326 block remained comparable with the rs3024493-unadjusted analysis, suggesting the associations from rs960326 block and rs1800890 block are independent. While the rs960326 block may indicate the presence of an independent functional variant in this region, none of these three SNPs passed the Bonferroni adjustments for multiple testing (table 3). Hence, the fine mapping analysis of 1q32.1 suggests the presence of at least two independent signals of association with melanoma OS, one of which (largely driven by rs3024493) associates with the significance level passing the multiple testing adjustments.
The associations of genetic variants from fine mapping analysis in 1q32.1 with melanoma overall survival
Putatively functional SNPs correlated with rs3024493

Fine mapping association analysis of 1q32.1 with melanoma overall survival. Twenty-four SNPs were selected from 1KGP and HapMap phase 3 with correlation of r2<0.6. The SNP associations with melanoma survival were tested individually for each variant using Cox regression models (Materials and methods) in multivariate analysis adjusted by age at diagnosis, gender, tumour stage, thickness, ulceration, and histological subtype. p Values were determined using 2 degrees of freedom test. (A) SNP-unadjusted analysis. (B) Analysis adjusted by the status of rs3024493. Circular markers represent SNPs chosen for fine mapping and square markers were part of the original tagging selection; the colour of the markers indicates the linkage disequilibrium (LD) correlation (r2) in relation to rs3024493, as displayed by legend in the upper right section of each plot.
Correlation of variants associated with melanoma OS at 1q32.1 with putatively functional SNPs from ENCODE data
None of the tagging SNPs associated with melanoma outcomes at 1q32.1 were located within gene coding regions. Using the data from the ENCODE project21 ,22 we tested whether the associated SNPs at 1q32.1 map in functionally important regulatory regions, providing potential functional mechanisms in melanoma progression. In fact, rs3024493, located in the third intron of IL10, maps within a DNAseI hypersensitivity cluster, multiple histone marks (H3K4Me1 and H3K27Ac), and binding sites for STAT3, NFKB, POLR2A. Also, less than 500 bp upstream from IL20, rs1400986 maps within histone marks (H3K4Me1, H3K4Me3, H3K27Ac) and binding sites for several transcription factors. In addition, using the low pass whole genome sequencing data from the 1000 Genomes Project (1KGP) we identified variants that correlate (r2>0.7) with the SNPs from the associated region at 1q32.1. In particular, as listed in table 4, using the ENCODE data we have found four proxies correlated with rs3024493, our most significant SNP, that map within putative transcription binding sites, DNAseI sites and/or histone activating regions.
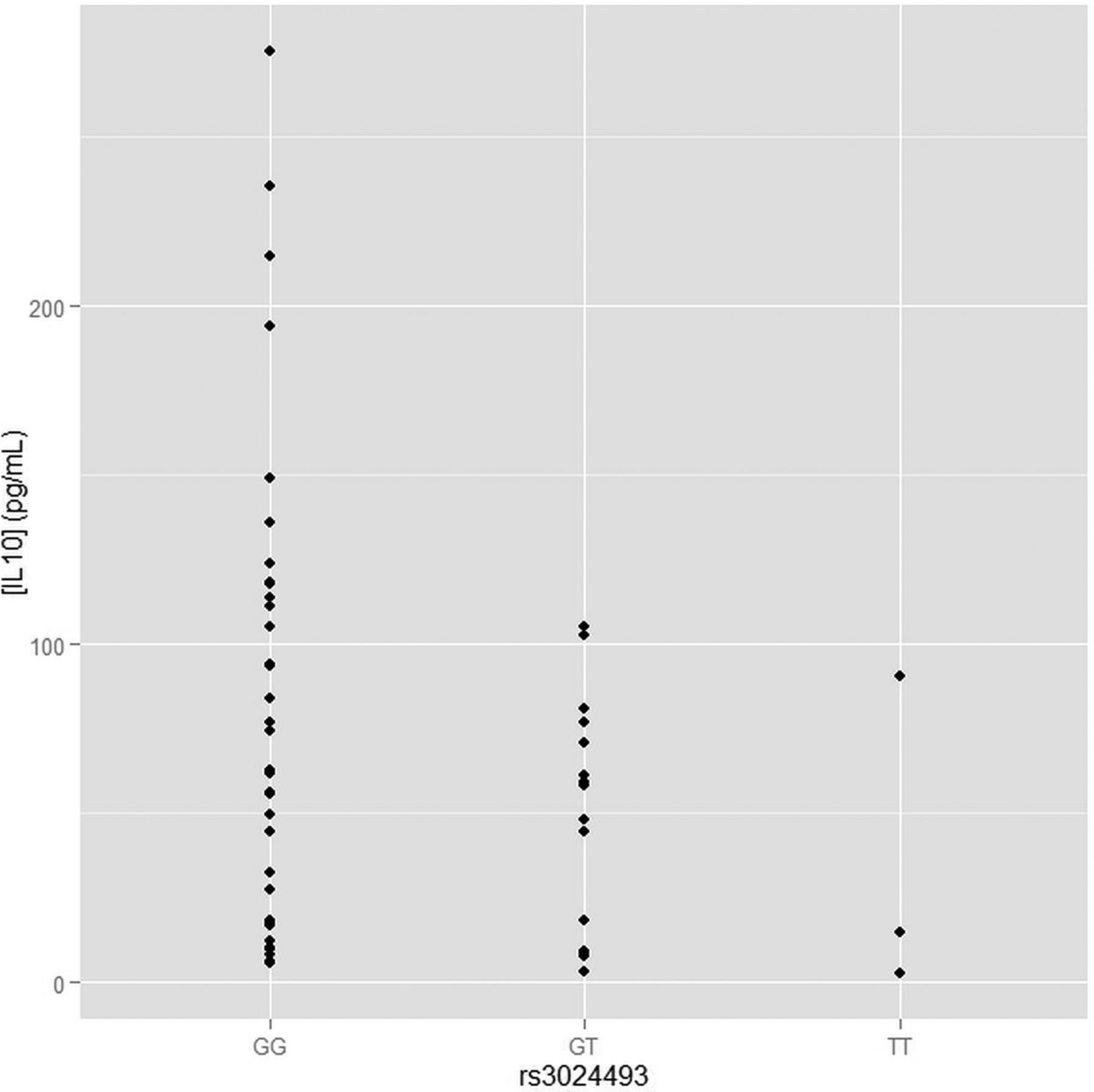
Correlation of rs3024493 with the secretion of IL10 in activated CD4+ T cells from melanoma patients
As rs3024493 is the strongest association found in the study, and the only association that has passed the Bonferroni adjustments for multiple testing, we have tested whether rs3024493 associates with the production of IL10 and six other related cytokines (IL2, IL4, IL6, IL17a, IFN, TNF). We examined the secretion of these immunomodulatory proteins by CD4+ T cells isolated from the peripheral blood of a subset of 75 melanoma patients selected from the population genotyped in the study. Following isolation of CD4+ T cells and activation by CD3/CD28 beads, cytokine secretion was quantified and compared between the three genotypes of rs3024493 (two degrees of freedom test) as well as in a dominant model (cytokine secretion compared between major allele homozygotes, as a reference, and aggregated heterozygotes and minor allele homozygotes). As shown in figure 2, after overnight stimulation, a statistically significant difference in IL10 secretion was observed in an allele-specific manner (Spearman correlation test p=0.025). IL10 secretion decreases with the dosage of minor allele, showing the lowest secretions for minor allele homozygotes. Because of the low minor allele frequency for this variant (0.08), we also performed dominant model analysis (supplementary figure S3), which revealed significantly decreased IL10 secretion in minor allele carriers (TT+GT, p=0.02) compared to major allele homozygotes (GG). For six other cytokines tested, the association between rs3024493 and cytokine production was not statistically significant (see online supplementary figure S3), suggesting that IL10 secretion is the only significant correlation with rs3024493 among the seven cytokines tested. Other variants from 1p32 associated with nominal significance with OS (in table 3) were also tested for IL10 secretion, but none of them were significant (see online supplementary figure S4).
{kind=link}
{kind=link}
Analysis of secretion of IL10 in CD4+ T cell stratified by the genotype of rs3024493. The secretion of IL10 was measured in isolated CD4+ T cells from a subset of 75 melanoma patients genotyped in the current study. Y axis represents the absolute concentration of IL10 (pg/mL), X axis shows the different rs3024493 genotype subgroup comparisons. Spearman correlation test was used for assessing the statistical significance between IL10 secretion and rs3024493 genotypes. The p value for genotype model (comparing the correlation between GG, GT and TT) was 0.02. Under dominant model (comparing GG vs GT+TT) we observed p=0.02 (see online supplementary figure S3).
Discussion
Somatic immune indicators or molecular and cellular factors of immune response have been associated with melanoma progression,9 ,23 ,24 indicating that the balance of the immune repertoire (including tumour and effector T cell response, TILs or cytokine profiles) is important in modulating melanoma clinical outcomes. We hypothesise that genetic variation in the genes related to these processes may affect the immune capacity of the host. Germline variants may represent efficient and easily assessable markers for more personalised clinical assessments, which is important considering the current limitations of accurate prognostication for melanoma patients at early, non-metastatic stages.
In our study we have confirmed statistically significant associations with melanoma OS for multiple germline variants in the locus of 1q32.1, which contains four interleukin genes (IL10, 1L19, IL20, and IL24). The strongest association signals stem from a relatively narrow region surrounding IL10. Several previous small studies have suggested a correlation between genetic variants in IL10 and melanoma OS.11 ,15 ,25 However, this promising evidence suffered from the lack of independent validation in a larger cohort, as well as the relatively narrow selection of candidate SNPs, complicating broader interpretation of these results. Specifically, the IL10 promoter SNP rs1800896 (−1082 G/A) has been consistently associated with overall melanoma survival in previous smaller studies; carriers of the minor allele (AG or GG) having increased survival time compared to major allele homozygote genotypes (AA).11 In our study, we have replicated the association of rs1800896 with melanoma survival observed in these prior reports in a much larger sample population. However, the strongest association in our scan comes from rs3024493, which surpasses adjustment for multiple testing and was not previously reported. Although the LD correlation between rs3024493 and rs1800896 is relatively low, it is possible that rs3024493 drives the associations found at 1q32.1, in particular by heterozygotes as we found here (HR 0.58, p=0.00069). While it is possible that the low MAF (0.16) for this variant in our cohort reduces the detection power of true effect for minor allele homozygotes, it is also likely that the protective association observed for rs3024493 heterozygotes may be due to the underlying genetic structure of the region and/or yet unexplained biological mechanisms related to the complex role of IL10 in immune response.
IL10 has traditionally been considered an immunosuppressive cytokine, exhibiting an anti-inflammatory response.26 Higher expression of IL10 in tumours has previously been associated with more aggressive tumor progression (invasive primary and metastatic melanoma) that correlated with IL10 expression in infiltrating lymphocytes and macrophages.27 Similarly, high IL10 serum values are consistently found to be associated with poor survival and advanced stages of the disease.27 ,28 Based on this and other evidence, the generally accepted model has postulated that increased expression of IL10 is implicated in inhibition of the anti-tumour T cell response in melanoma due to downregulation of T cell function, resulting in tumour escape. Although in our study we have tested circulating T cells and not TILs, this trend is also apparent in our data where we found that decreased secretion of IL10 by CD4+ T cells significantly correlated with the dosage of the minor allele (T allele) of rs3024493. However, the stronger association of rs3024493 heterozygotes with survival found in our study may lead to an intriguing conclusion that mid-low levels of IL10 (secreted by GT heterozygotes) correlate with better prognosis, while both the high levels (by GG reference homozygotes) and low levels (by TT minor allele homozygotes) of IL10 correlate with worse outcome, suggesting that IL10 exerts both anti- and pro-tumoral activities. This is supported by recent groundbreaking research revealing that IL10 is highly pleiotropic and may actually exhibit immune-mediated tumour rejection properties.29–31 Specifically, these recent observations demonstrated that through interactions with other important immune molecules, IL10 induces several essential mechanisms for effective antitumour immune surveillance such as inducing infiltration and activation of intratumoral cytotoxic T cells or triggering the expression of other cytokines such as IFNγ. Our observations that GT heterozygotes, as ‘mid-secretors’ of IL10, manifest with protective effect (better outcome) while TT homozygotes, as ‘low-secretors’ of IL10, show strong risk effect (worse outcome), suggest that the pro-stimulatory immune response against melanoma is most likely determined by the optimal balance of IL10 secretion. While such a model is possible and supported by our findings, further experimental elucidation will be needed to link IL10 genetic variants with IL10 antitumour response. As part of this subsequent analysis, it would be important to consider the origin of cells secreting IL10 in tumour microenvironment or in circulation as this may further determine potential IL10-driven anti- or pro-tumoral effects. For example, IL10 also induces Tregs that can be both promoting and preventing tumour rejection, and since our analysis only tested broad spectra of CD4+ T cells (including Tregs), further sub-classification of T cell populations will be critical in estimating the effect of observed genetic associations on IL10 secretion in T cell subtypes.
The presence of other yet-unknown genetic variants with less common or rare frequencies may also be driving the associations observed in this study. In our haplotype analysis (see online supplementary table S5), we noted that among our patient cohort the minor allele of rs3024493 tags only one haplotype TTTGA (constituted by rs3024493, rs3024491, rs2222202, rs1800896, rs1800890), and while one copy of this haplotype provides a survival benefit, two copies significantly decrease survival. This is in accordance with the pleiotropic model of IL10/rs3024493 secretion proposed herein, further supporting the biological mechanisms behind reverse associations for rs3024493. The less common frequency of this haplotype in our patient cohort (MAF=0.02) may indicate the presence of rare, yet-unknown coding or regulatory variants associated with this haplotype potentially impacting IL10 secretion. The detailed re-sequencing of this locus in patient carriers of this haplotype would hence be needed to test such a possibility. The other common variants correlated with rs3024493 may also provide important insight into the biological mechanism behind these observations. For example, in a recent study investigating the role of rs3024493 in IL10 in systemic lupus erythematosus (SLE), one proxy SNP, rs3122605, was shown to create a novel binding site for the transcription factor Elk-, correlating with increased expression of IL10 in peripheral blood lymphocytes.32 The association between rs3024493 and melanoma survival in our study may be driven by this same effect; however, as we show, rs3024493 and four other highly correlated SNPs are located within a region comprised of multiple transcription factor binding sites and histone marks (table 4). In fact, the narrow region of ∼16 kb represented by these five highly correlated SNPs seems to map in the transcriptional regulatory hotspot, which may explain the biological mechanisms underlying the observed associations of IL10 variants with melanoma survival and which will need to be functionally interrogated in greater detail.
Interestingly, rs3024493 and other SNPs in 1q32.1 were identified in previous genome-wide association studies (GWAS) on multiple autoimmune disorders, including SLE and ulcerative colitis, further supporting an important clinical impact of this variant on IL10 in immune regulation.33–36 In fact, a recent report on juvenile idiopathic arthritis, a rare young-at-onset autoimmune disease,37 shows multiple SNP associations at 1q32.1 including rs1400986 (from rs960326 block as defined in our study). Similarly, rs1800871, which correlates with the rs1800890 block at 1q32.1, was the most significant association in a recent GWAS on Behçet’s disease.38 The functional analyses in many of these reports confirmed the low production of IL10 associated with minor allele, consistent with the trend found in our study. As autoimmunity is a manifestation of the up-regulation of the immune response, it is possible that the autoimmune traits may be linked to the attenuation of melanoma progression and therefore better outcome in melanoma patients. While our association data in the IL10 locus at 1q32.1 strongly support such a scenario, this needs to be systematically tested in a separate study conditioning on the status of rs3024493 and other observed associations in our scan.
Interestingly, we observed relative discordance between variants that were significantly associated with OS and RFS. The statistical power of RFS versus OS analyses may play an important role in these differences. The Cox model, applied here, usually has reduced statistical power to detect significant associations with RFS due to shorter time periods measured as opposed to OS analysis where the observation periods are much longer. This suggests that the significant associations observed with OS may be undetected in RFS analysis. However, we have observed a strong positive correlation between OS and RFS for OS-associated variants when considering HRs instead of significance level (Pearson’s coefficient (r)=0.89, p<0.0001; see online supplementary figure S5). Importantly, the rs3024493 genotype-dependent association effects found significant for OS clearly correlate with the effects for this SNP observed for RFS (protective effect for GT, risk effect for TT). This clearly supports the consistency of our most significant result in both clinical end-points and hence the plausibility of pleiotropic effects of IL10/rs3024493 in modulating the melanoma outcomes. While these concordant correlations are supportive, there are other potential survival impacting factors that were not possible to account for in our analysis. Among these, adjustments by treatment outcomes or the assessment of melanoma-specific mortality should be considered in the future expanded design.
It is also possible that the observed associations may be impacted by particular CM subtypes. For example, acral melanomas (AM) manifest with significantly worse survival rates compared to CM.39 Since these occur predominantly in other non-Caucasian ancestries it was not possible to examine AM separately in our study, as these were underrepresented (N=26). However, although the adjustment of our data by anatomical location (extremities, trunk, acral) did not significantly alter the association results (data not shown), the assessment of immune pathways contributing to the clinical outcomes in these different CM manifestations will be important in the future focused analyses.
In conclusion, our study provides strong support for a role of the interleukin locus at 1q32.1 in the modulation of melanoma survival. We provide evidence for a novel association with OS in the region of IL10 (rs3024493), which surpasses the significance level for multiple testing. In addition, we replicate the associations from previous small sized studies (rs1800896). We provide functional evidence showing that rs3024493 correlates with the secretion of IL10 in CD4+ cells, suggesting the putative role of this variant in immune-specific melanoma evasion. The association with rs3024493 found here suggests possible alternative mechanisms by which IL10, traditionally thought to exert immune-suppressive properties, contributes to immune-stimulatory tumour rejection. Lastly, our findings provide an intriguing link between melanoma clinical outcomes and autoimmunity, as the most significantly associated SNPs in our study were among the top loci associated with the risk of several autoimmune traits in recent GWAS. While the current study already represents an independent validation in a large melanoma patient cohort, a more comprehensive functional analysis of all interleukin genes in the region will need to be performed. Also, the detailed re-sequencing of the locus may uncover less common or rare variants within the same haploblocks found to be associated with melanoma outcomes in our study. In addition, the investigation between associated variants and other immune indicators of tumour microenvironment (eg, tumour-infiltrating lymphocytes) will be important as these were recently suggested as biomarkers of melanoma survival.10 These additional suggested investigations may prove crucial in clarifying the ambiguity surrounding the role of specific interleukins from 1q32.1 in tumour immunity. At the same time, it is likely that the data from this and subsequent studies will provide solid support for the consideration of 1q32.1 locus as a clinically relevant biomarker that could guide patient management in terms of follow-up and treatment.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors JR, MV, MK, TK designed the study. JR, MV, DM, MK, TK drafted the manuscript. JR, MV, AB, CA, IS, DM, JS performed experiments. JR, MV, DM, CNM, YS, MK, TK performed analyses. RS, RB, AP, DP, IO, MK, TK provided material and reagents for the study. JR, MV, DM, CNM, NF, DP, IO, MK, TK provided critical review of the manuscript. RS, IO, TK provided financial support for the study.
-
Funding The study was funded by the grants from National Cancer Institute 1R21CA184924-01 (TK) and Cancer Center Support Grant P30CA016087.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Institutional Review Board NYULMC.
-
Provenance and peer review Not commissioned; externally peer reviewed.