Article Text

Abstract
Background Coenzyme Q is an essential mitochondrial electron carrier, redox cofactor and a potent antioxidant in the majority of cellular membranes. Coenzyme Q deficiency has been associated with a range of metabolic diseases, as well as with some drug treatments and ageing.
Methods We used whole exome sequencing (WES) to investigate patients with inherited metabolic diseases and applied a novel ultra-pressure liquid chromatography—mass spectrometry approach to measure coenzyme Q in patient samples.
Results We identified a homozygous missense mutation in the COQ7 gene in a patient with complex mitochondrial deficiency, resulting in severely reduced coenzyme Q levels We demonstrate that the coenzyme Q analogue 2,4-dihydroxybensoic acid (2,4DHB) was able to specifically bypass the COQ7 deficiency, increase cellular coenzyme Q levels and rescue the biochemical defect in patient fibroblasts.
Conclusion We report the first patient with primary coenzyme Q deficiency due to a homozygous COQ7 mutation and a potentially beneficial treatment using 2,4DHB.
- Metabolic disorders
- Molecular genetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Coenzyme Q (CoQ), also known as ubiquinone, is a small fat-soluble redox cofactor that has been extensively studied for its essential role as a mitochondrial respiratory chain electron carrier. It consists of a redox-active benzoquinone ring and a polyisoprene tail. The tail is believed to be important for diffusion in the lipid bilayer, as well as for interactions with ubiquinone redox enzymes,1 and comprises 6 units in yeast (CoQ6), 9 units in rodents (CoQ9) and 10 in humans (CoQ10). Aerobic respiration requires that electrons from energy-rich intermediates such as NADH or FADH2 be passed to molecular oxygen, via the respiratory chain in the inner mitochondrial membrane. CoQ is essential in this process, shuttling electrons from NADH dehydrogenase (complex I) or succinate dehydrogenase (complex II) to the cytochrome bc1 complex (complex III). Although predominantly located in the inner mitochondrial membrane, CoQ is present in almost all cellular membranes in aerobic organisms.2 In addition to its role as an electron shuttle during aerobic respiration, CoQ is also a cofactor of various uncoupling proteins and other mitochondrial dehydrogenases, and functions in lysosomes, endoplasmic reticulum and plasma membranes. Furthermore, CoQ is the only lipid-soluble antioxidant synthesised entirely by animal cells.2
CoQ10 levels naturally diminish with age,2 but can also be decreased due to treatments with, for example, statins due to a shared biosynthesis pathway with cholesterol.3 Additionally, CoQ10 deficiencies (MIM 607426, 614651, 614652, 612016, 614654, 614650) have been reported in a range of patients with inborn errors of metabolism,4 often identified by severe mitochondrial dysfunction with combined deficiencies of the respiratory chain complexes. They can be caused by either a primary defect in the CoQ10 biosynthetic pathway or as a secondary phenomenon in a range of dysfunctions for yet unclear reasons.
Despite its central role in aerobic respiration, the biosynthesis of CoQ is not fully understood. Based on studies in yeast, the products of at least nine genes, Coq1–Coq9, are known to be involved in CoQ synthesis, which is initiated by the condensation of the isoprenoid tail with the benzoquinone ring precursor.5 A series of benzoquinone ring modifications, including C-hydroxylations, decarboxylations, O-methylations and C-methylations, subsequently result in CoQ10 formation. For some of the corresponding proteins (COQ1, COQ2, COQ6, COQ3, COQ5, COQ7), the biochemical functions are understood, whereas for others, the mechanisms behind their involvement in the pathway remain unclear (COQ4, COQ8, COQ9).
Mutations in PDSS1, PDSS2, COQ2, COQ4, COQ6, COQ9, ADCK3 and ADCK4 are known causes of primary CoQ deficiencies in humans.3 PDSS1 and PDSS2 form a heterotetramer, performing the analogous function to Coq1 in yeast, whereas ADCK3 is the mammalian Coq8 counterpart. ADCK4 is a paralog of ADCK3 and has been shown to interact with members of the CoQ biosynthesis pathway, including COQ5,6 COQ6 and COQ7.7 COQ7 has further been shown to interact with COQ9,8 suggesting the presence of a large CoQ10 biosynthesis complex.
Materials and methods
Biochemistry
Mitochondrial ATP production rates (MAPRs) and respiratory chain enzyme activities in skeletal muscle and mitochondrial oxygen consumption of fibroblasts were determined as described in online supplementary information.
Next generation sequencing, MIP analysis and Sanger sequencing
Whole exome sequencing was performed on DNA from the patient and parents’ fibroblasts, using Illumina technology. Following mutation identification pipeline (MIP) analysis, among the top 20 variants only the c.422T>A transition in COQ7 assumed homozygosity and a mitochondrial association. Sanger sequencing was performed on genomic DNA extracted from blood from the patient, unaffected sibling and both parents (for details see ref. 9 and online supplementary information).
Measuring absolute ubiquinone levels
Ubiquinone quantification by ultra-pressure liquid chromatography (UPLC)—tandem quadrupole mass spectrometry analysis was performed as described in online supplementary information.
Results
In this study, we report the first case of COQ7 deficiency. The affected boy was born in 2005 as the second child of consanguineous Syrian parents, with an older healthy sister born in 2002. Pregnancy was complicated by oligohydramniosis, fetal lung hypoplasia and growth retardation. The boy was born full term but small for gestational age (birth weight 2070 g, length 46 cm, head circumference 30.5 cm). At birth, he had muscular hypotonia, contractures of the extremities and respiratory distress with persistent pulmonary hypertension of the newborn. His Apgar score was 1,5,7. Lung hypoplasia was confirmed and renal dysfunction was diagnosed, with plasma creatinine elevated up to 196 µmol/L (ref <100) on day 2. Ultrasound revealed small dysplastic kidneys with impaired cortical differentiation. Secondary, there was systemic hypertension and left ventricular cardiac hypertrophy. Plasma creatinine normalised during the first week. Blood pressure normalised after 3 months, the cardiac hypertrophy regressed and pulmonary function normalised within the first 8 months. Follow-up ultrasound of the kidneys has shown normal growth and appearance, and there are no signs of glomerular or tubular dysfunction so far. A moderate developmental retardation was seen; the boy learned to sit without support at 10–11 months and crawled reciprocally after 1 year of age. He never learned to stand or walk independently. Electrophysiological investigations at 2 years of age showed peripheral sensorimotor polyneuropathy of axonal and demyelinating type. An MRI of the brain performed 2 months later was normal. Postnatal growth continued to be retarded; weight following −3 SD curve, height −2 SD and head circumference below −3 SD. Due to persistent feeding difficulties, he received a gastrostomy at the age of 2 years and 10 months. His weight subsequently improved but he remains short for his age. The patient is currently 9 years old with mild learning disabilities, hearing impairment due to combined sensorineural and conduction defects, and visual dysfunction. His musculature is thin and there is significant muscular hypotonia. The muscle weakness has been progressive with joint displacements and pain; the boy is no longer able to sit. The patient was initially treated with idebenone, but after the diagnosis of a primary CoQ10 deficiency he was placed on CoQ10 treatment, stalling the regression and significantly reducing the pain.
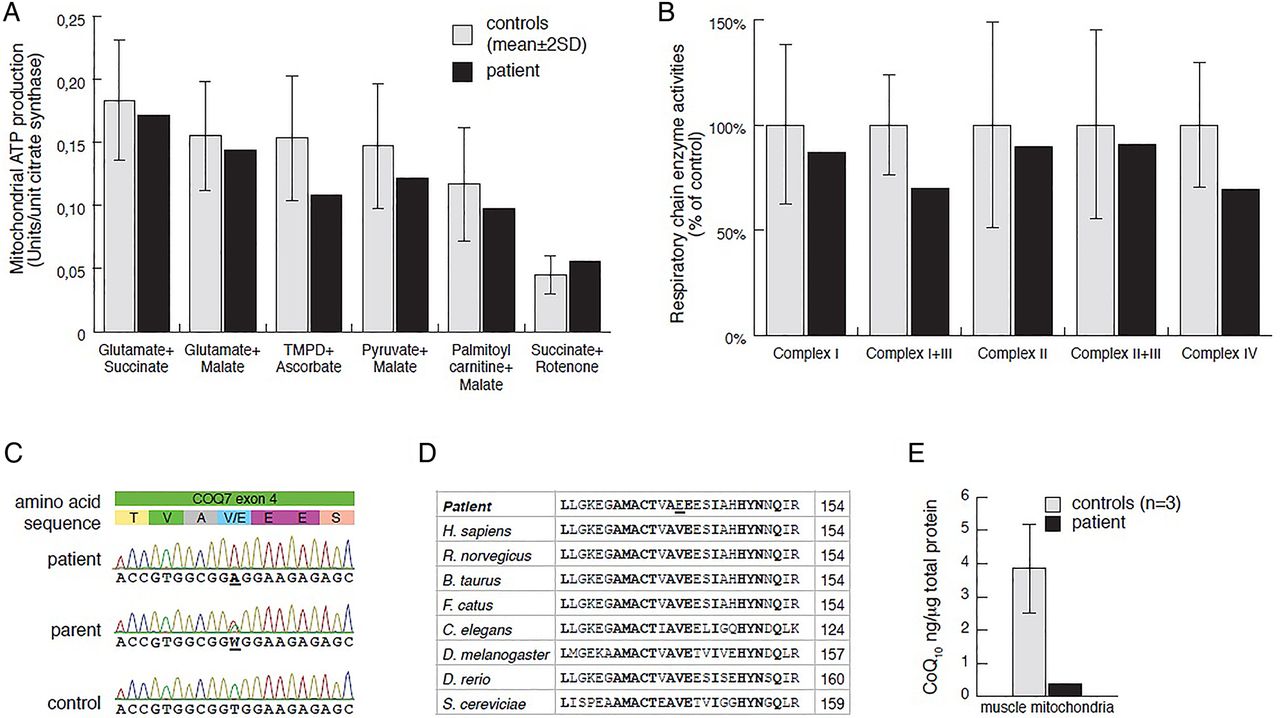
Laboratory investigations at 2 years of age showed slightly elevated plasma lactate (3.5 mmol/L, ref 0.5–2.3), cerebrospinal fluid (CSF) lactate (4.4 mmol/L, ref 0.5–2.3) and increased CSF albumin to 652 mg/L (ref <225). Urine analysis revealed moderately increased excretion of fumarate and malate. A muscle biopsy was performed at 2 years 3 months to measure MAPR and respiratory chain enzyme activities (see online supplementary material and methods), revealing a combined complex I+III and IV deficiency (figure 1A, B). Histological examination of the biopsy showed small fibre size indicating neurogenic damage but no obvious myopathic changes. Electron microscopy revealed no obvious abnormalities of mitochondria or other structures. Sanger sequencing of the complete mtDNA genome and the POLG gene revealed no pathogenic mutations.

Mitochondrial respiration and respiratory chain enzyme activities in skeletal muscle, results from Sanger sequencing, and CoQ10 levels. (A) Mitochondrial ATP production rate was determined by a firefly luciferase-based method at 25°C, using six different substrate combinations as indicated. Results are presented as the ATP synthesis rate (units) per unit of citrate synthase (CS) activity (control n=11; age 0–5 years). (C) Respiratory chain enzyme activities of complexes I and III (NADH:cytochrome c reductase), complex I (NADH:coenzyme Q reductase), complexes II and III (succinate:cytochrome c reductase, SCR), complex IV (COX) and CS were determined. Results are presented as percentage of mean control (n=9; age 0–5 years) values. The range of control values is depicted as ±2SD. (C) Electropherograms showing patient, one parent and a control sample for COQ7 exon 4. (D) Sequence alignment of eight representative eukaryotic COQ7 peptide sequences (Geneious R6 software by Biomatters (http://www.geneious.com)), indicating conserved sites (bold) and the p.Val141Glu patient mutation (underlined). Amino acid position is indicated in the right column. (E) CoQ10 levels in mitochondrial extracts from skeletal muscle were determined in patient (black) and control (grey; n=3) samples, using an Acquity UPLC (Waters) connected to a XevoTM TQ (Waters) (for details see the Materials and methods section and online supplementary information).
We performed whole exome sequencing on genomic DNA samples from the patient and his parents, followed by in-house computational analysis, using the MIP9 ,10 (see online supplementary information). After MIP analysis, only three candidate genes assumed an autosomal recessive inheritance of potentially disease-causing variants and only a single candidate gene was associated with mitochondria. Other candidates were dismissed for failure to show an appropriate inheritance model. Sanger sequencing confirmed (see online supplementary material and methods) homozygosity for a thymidine to adenosine transversion at nucleotide 422 of COQ7 (MIM 601683; NM_016138:exon4:c.422T>A:p.Val141Glu) in the patient and heterozygosity in both parents and the unaffected sibling (figure 1C). The variant was not present in any of the public databases or in our in-house database containing data from 156 individuals.
COQ7 is a di-iron oxidase responsible for the penultimate step of CoQ synthesis, hydroxylating 5-demethoxyubiquinol (DMQH2) in the presence of NADH.11 ,12 The highly conserved glutamic acid p.Glu142 residue is predicted to be part of the di-iron motif,13 and with p.Val141 being conserved in eukaryotes (figure 1D), it is highly likely that the p.Val141Glu mutation affects COQ7 function and impairs iron binding.
To identify whether CoQ levels are affected in the patient, we developed a novel method to analyse CoQ10 levels in isolated mitochondria, using UPLC—tandem quadrupole mass spectrometry analysis (see online supplementary material and methods). In our analysis, this method was highly sensitive to even low levels of input sample and CoQ10 concentrations, and we were able to identify a severe reduction in CoQ10 levels in mitochondrial extracts from patient skeletal muscle samples in comparison with controls, strongly supporting that the p.Val141Glu substitution impairs CoQ10 biosynthesis (figure 1E).
Previous studies in yeast demonstrated that benzoic acid derivatives are capable of bypassing certain primary CoQ10 biosynthesis deficiencies.14 ,15 Specifically, the resorcylic acid, 2,4-dihydroxybenzoic acid (2,4-dHB) is capable of bypassing the enzymatic step performed by Coq7 in a Coq7 knockout (KO) yeast strain, overexpressing Coq8,15 and has recently been shown to rescue COQ7 KO mice.16 In agreement with our observations made in skeletal muscle mitochondria, patient fibroblasts also displayed reduced CoQ10 levels, both in total cell extracts, as well as mitochondrial preparations (figure 2A). Supplementation of the culture media with 0.1 mM 2,4-dHB resulted in increased CoQ10 levels after 7 days, which could be reversed after additional 7 days incubation in the absence of 2,4-dHB (figure 2B). Surprisingly, CoQ10 concentrations decreased in treated control cells, suggesting that 2,4-dHB might compete with DMQH2 for COQ7 activity. Measuring mitochondrial respiratory chain enzyme activities revealed a clear combined complex I+III and II+III deficiency in patient fibroblasts, while cells grown in the presence of 2,4-dHB had improved complex I+III and II+III activities, with predominantly the former benefiting from the treatment (figure 2C). We further assessed mitochondrial respiration in permeabilised cells. Patient fibroblasts presented with reduced oxygen consumption, both with complex I, as well as complex I and II substrates, and the addition of the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazone revealed only 40% of maximal respiration in patient fibroblasts in comparison with controls (figure 2D). In contrast, in the presence of 2,4-dHB, mitochondrial respiration was fully restored in patient fibroblasts (figure 2D), showing a complete rescue of the phenotype and indicating a potential treatment strategy of the COQ7 defect. Finally, to further confirm pathogenicity of the p.Val141Glu substitution, we transiently expressed human COQ7 in patient fibroblasts, resulting in improved mitochondrial respiration (figure 2E). Transfection efficiency was estimated to approach more than 80% by coexpression of eGFP in transfected cells.

{kind=link}
{kind=link}
CoQ10 levels, mitochondrial respiratory chain activities and respiration in fibroblasts. (A) CoQ10 levels in mitochondrial and total fibroblast extracts were determined in patient (black) and control (grey; n=3) samples as described above. (B) CoQ10 levels in control (n=3) and patient fibroblasts in the presence or absence of 2,4-dihydroxybenzoic acid (2,4-dHB) treatment. Shown is the average of two independent experiments. (C) Respiratory chain enzyme activities of complexes I and III (NADH:cytochrome c reductase), complex I (NADH:coenzyme Q reductase), complexes II and III (succinate:cytochrome c reductase, SCR), complex IV (COX) and citrate synthase (CS) were determined in mitochondrial extracts from control (grey) and patient (black) fibroblasts. Cells were grown in high (4.5 g/L) glucose, Dulbecco's modified Eagle's medium, 10% fetal bovine serum and pyruvate in the presence (light grey, black) or absence (dark grey or white) of 0.1 mM 2,4-dHB. (D) Mitochondrial oxygen consumption of the patient (black and white) or controls (light and dark grey) in the presence or absence of 2,4-dHB. Measurements were performed in the presence of the complex I substrates glutamate, malate, pyruvate (GMP) and ADP; complex I+II substrates GMP, succinate and ADP, or GMP, ADP, succinate and the mitochondrial uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP). Error bars indicate the SE between three independent experiments. (E) Mitochondrial oxygen consumption of patient (black, white) or control (light and dark grey) fibroblasts transiently transfected with pIRES2-eGFP_hCOQ7 or empty vector. Shown is the average of two independent experiments.
Human CoQ10 deficiencies are associated with a heterogeneous spectrum of clinical phenotypes,3 ,17 including an infantile multiorgan disease (COQ2), encephalopathy (PDSS1, PDSS2, COQ2, COQ4), psychomotor delay (COQ2, COQ4, ADCK3, COQ9), stroke-like lesions (COQ2, ADCK3), dystonia (COQ2, ADCK3), hypotonia (COQ2, COQ4, ADCK3), cerebellar atrophy and ataxia (COQ2, COQ6, ADCK3), epilepsy (COQ2, COQ6, ADCK3, COQ9), feeding problems (COQ2), deafness (PDSS1, COQ6), optic atrophy (COQ2), axonal neuropathy (PDSS1, ADCK3), myopathy (COQ2, COQ4), dysmorphic features (COQ4, COQ6), exercise intolerance (ADCK3), nephropathy (PDSS1, PDSS2, COQ2, COQ6, ADCK4) and tubulopathy (COQ9). Mitochondrial respiratory chain deficiencies are notoriously heterogeneous but some of the above features may be more specific for the primary deficiencies of CoQ, possibly reflecting additional functions of this molecule. In particular, steroid-resistant nephrotic syndrome is a prominent presentation in patients with COQ2,18 COQ6,19 ADCK47 mutations, and the single patient described with COQ9 deficiency also had renal dysfunction.20 The renal phenotype was studied in detail in cells from patients with mutations in ADCK4, demonstrating reduced CoQ10 levels and reduced mitochondrial respiratory enzyme activity.7 Additionally, a causative association between ubiquinone and renal dysfunction was shown in knockdown experiments of Adck4 in zebrafish and Drosophila,7 as well as mice with a spontaneous missense mutation in Pdss2,21–25 strongly suggesting that kidney function is highly sensitive to CoQ levels.
The patient described here has a COQ7 deficiency with a complex clinical picture and multiple organ involvement. His neonatal lung hypoplasia, contractures, early infantile hypertension and cardiac hypertrophy, however, are likely secondary to his prenatal kidney dysplasia with renal dysfunction and resulting oligohydramniosis. Remarkably, this resolved during his first year, and kidney growth and renal function were normal, even prior to the initial idebenone treatment. Idebenone treatment has been proposed to be inefficient in primary deficiencies of CoQ,26 ,27 and the patient's neurological symptoms remained progressive, especially his peripheral neuropathy with resulting muscular hypotonia and atrophy. He also suffers from mild psychomotor delay with hearing and visual impairment. However, upon diagnosis the patient was placed on CoQ10 treatment, which seems to have stabilised disease progression.
In conclusion, we present the first report of CoQ10 deficiency caused by a homozygous mutation in the gene encoding the ubiquinone biosynthesis protein COQ7. The unusual clinical picture, with prenatal onset kidney dysplasia and renal dysfunction that normalised during the first year, highlights the importance of CoQ10 for kidney development and function. It also emphasises that the requirement for CoQ10 varies between tissues and developmental stages, and indicates that it is dynamic and possibly affected by supplementation therapy. Our results suggest that CoQ10 biosynthesis is not linear, but that CoQ10 metabolites can enter at several different stages of the pathway, suggesting that like in yeast14 ,15 ,28 resorcylic acid analogues might be useful in the identification of primary CoQ10 deficiencies and provide an alternative treatment strategy for patients suffering from CoQ10 deficiencies. Support stems from the recent report that COQ7 deficient mice could be successfully treated with 2,4-dHB, while CoQ10 supplementation had little to no effect on the animals.16
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
CF, HS and KN contributed equally to this work.
Contributors Experimental design and execution: CF, NL, AM, YV, HB, AF, CM, MB, RW, AW, AWe. Manuscript preparation: CF, AW, AWe. Data analysis: CF, HS, KN, AM, AF, NL, HB, ME, RW, MB, MM, RA, RHZ, UvD, AW, AWe. Clinical investigation: KN, ME, RHZ, UvD, AW, AWe. All authors have read and understood the final manuscript.
Funding This work was supported by the Swedish Research Council (12198), the Karolinska Institutet, the Stockholm County Council, the Swedish Brain Foundation, and the Knut and Alice Wallenberg Foundation. AWr is a Ragnar Söderberg fellow (M77/13) and is supported by the Swedish foundation for strategic research (SSF) (4-1247/2013) and the Swedish research council (young investigators) (521-2012-2571).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Regional ethics committee at Karolinska Institutet.
Provenance and peer review Not commissioned; externally peer reviewed.