Article Text

Abstract
Background Craniosynostosis (CRS) is a premature closure of calvarial sutures caused by gene mutation or environmental factors or interaction between the two. Only a small proportion of non-syndromic CRS (NSC) patients have a known genetic cause, and thus, it would be meaningful to search for a causative gene disruption for the development NSC. We applied a whole genome sequencing approach on a 15-month-old boy with sagittal and metopic synostosis to identify a gene responsible for the development of the disease.
Methods and results Conventional chromosome study revealed a complex paracentric inversion involving 2q14.3 and 2q34. Array comparative genomic hybridisation did not show any copy number variation. Multicolour banding analysis was carried out and the breakpoints were refined to 2q14 and 2q34. An intronic break of the PTH2R gene was detected by whole genome sequencing and fluorescence in situ hybridisation analysis confirmed disruption of PTH2R.
Conclusions We report PTH2R as a gene that is disrupted in NSC. The disruption of the PTH2R gene may cause uncontrolled proliferation and differentiation of chondrocytes, which in turn results in premature closure of sutures. This addition of PTH2R to the list of genes associated with NSC expands our understanding of the development of NSC.
- Chromosomal
- Clinical genetics
- Developmental
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Craniosynostosis (CRS) is defined by the premature osseous obliteration of one or more of the cranial vault sutures. The disruption of a fine balance between proliferation and differentiation during embryogenesis or early childhood can alter the patency of these sutures. Non-syndromic CRS (NSC) cases, which occur as an isolated anomaly, account for >80% of all CRS cases and most commonly affect sagittal sutures.1 Although multiple reports have identified mutations in several genes such as FGFR1-3, TWIST1/2, MSX2, FGFRL1, SNAIL, SLUG, NELL1 and RUNX2 in NSC, only a small proportion of patients have a known genetic cause, which indicates that in addition to strong genetic components the premature closure of the sutures is a complex trait.1 Recently, in an attempt to find a genetic cause of NSC, a locus near BMP2 and within BBS9 was implicated in the association of non-syndromic sagittal suture.2 Each sutural synostosis shows distinct characteristics, even among cases with known genetic components. Of note, chromosomal aberrations and submicroscopic chromosomal rearrangements have been associated more frequently with midline synostoses.3 Recent advances in whole genome sequencing (WGS) technologies have made it possible to identify genomic rearrangements and breakpoints involved in these rearrangements, facilitating rapid identification of disease genes in chromosomal breakpoint regions.4 Here, our objective is to identify a gene that is disrupted to cause the disease in a patient with midline NSC using cytogenetic analysis and WGS.
Materials and methods
Patient information
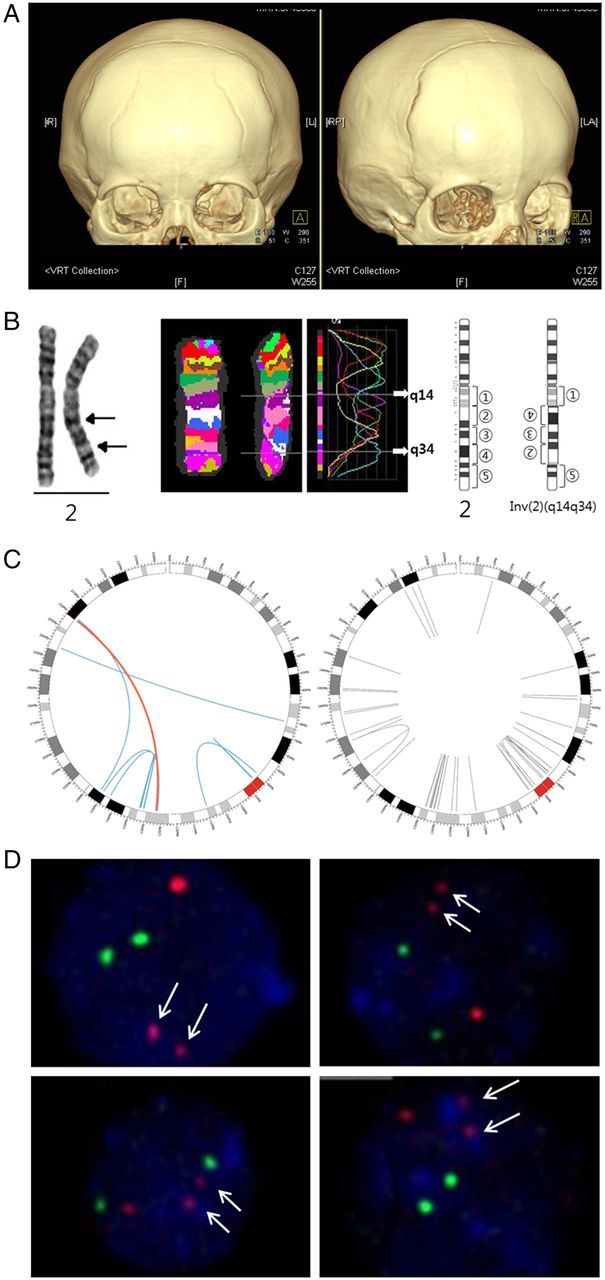
The patient was a 15-month-old boy, the first child of healthy non-consanguineous parents without any family history of CRS. He was born at 40 weeks gestation by caesarean section with normal birth parameters (weight of 3660 g). He presented with hearing impairment and delayed development. At the age of 15 months, clinical examination revealed hypertelorism, crumpled ear, bulging anterior fontanelle and premature closure of the coronal suture. Brain MRI suggested sequelae of previous periventricular white matter injury (periventricular leukomalacia). A three-dimensional facial bone CT scan showed synostosis of the sagittal and metopic sutures (figure 1A). His weight, height and head circumference at the age of 18 months were 9000 g, 78 cm and 41.5 cm (less than third centile), respectively. A craniofacial distraction osteogenesis was performed and followed up with postoperative helmet moulding therapy. Chromosomal study of peripheral blood using the G-banding with trypsin-Giemsa (GTG) technique at 550 band resolution revealed a complex paracentric inversion involving 2q14.3 and 2q34 (figure 1B). His parents and sister had normal karyotypes.

{kind=link}
PTH2R is a gene that is disrupted in craniosynostosis. (A) Three-dimensional CT scan showing a synostosis of the metopic and sagittal sutures. (B) Conventional chromosome analysis, which revealed a paracentric inversion involving 2q14.3 and 2q34 and multicolour banding (mBAND) analysis refined breakpoints to 2q14 and 2q34. G-banding (left end), multicolour banding (middle) and partial G-banded ideogram (right end) of chromosome 2 are shown. (C) Circos plot of eight inversions on chromosome 2 detected by analysis of whole genome sequence data. Left and right figures show inversions observed in the patient and in 22 normal controls, respectively. Curves indicate both boundaries of each inversion detected by analysis of whole genome sequence data; blue and red curves represent eight candidate inversions that passed multiple filters and grey curves represent other inversions (see online supplementary table S1 for details). The red curve represents the inversion that led to the intronic break of the PTH2R gene, which resides on 2q34, and that was not observed in normal controls. This intronic break was consistent with the breakpoints detected by mBAND analysis and was confirmed by fluorescence in situ hybridisation (FISH) analysis. (D) FISH analysis showed one brighter red signal and two small red ones (arrow), indicating a breakpoint within the PTH2R gene. Cohybridisation of green 5-fluorescein 2′-deoxyuridine 5′-triphosphate-labelled control probes, which hybridised to the centromeric region of chromosome 2, revealed two normal green signals. L, left; LA, left anterior; R, right; RP, right posterior; VRT, volume rendering technique.
Array comparative genomic hybridisation (CGH)
We considered local copy number (amplification and deletion) enriched regions using the NimbleGen CGX-12 array-chip (Roche NimbleGen, Wisconsin, USA) and the Agilent SurePrint G3 Human 1M Custom array CGH (Agilent Technologies Inc., California, USA), which was scanned using the Agilent Microarray Scanner, all according to the manufacturer's instructions. Copy number variations were detected by the ADM-2 algorithm in the Agilent Genomic Workbench Lite Edition 6.5.0.18.
Multicolour banding analysis
To better localise the breakpoints, multicolour banding (mBAND) analysis was carried out according to standard techniques and hybridised with a commercially available mBAND probe (MetaSystems, Altlussheim, Germany) for chromosome 2 (XCyte 2). Hybridisation, post-hybridisation washes and signal detection conditions were carried out following the method described by Tawn et al.5
Whole genome sequencing
We sequenced the entire genome of the patient using HiSeq 2000 (Illumina, California, USA) with paired reads of 101 base pairs (bp) and inserts of 500 bp. We checked the quality of the sequence reads using the FastQC 0.1 software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and trimmed the last 41 bp sequences from reads using the NGSQCToolkit 2.2.3 software.6 Using the BWA V.0.5.9 software, we aligned the paired-end reads to the human reference genome (February 2009 assembly of the human genome (hg19, GRCh37)).7 In addition, we also used the first Korean individual genome sequence (SJK) data as a reference genome.8
Detection and filtering of inversions
We extracted anomalously mapped paired-end reads using the SVDetect 0.8 software,9 including reads mapped on two different chromosomes with an incorrect strand orientation or pair order, or with an insert size longer than threefold of the SD from the mean. Based on calculation for one million reads, we set the mean insert size (µ) at 542.93 bp and the SD (σ) at 38.34 bp. We detected structural variations from the anomalously mapped reads using the BreakDancer max 1.1 software.10 The fixed window size used by BreakDancer is defined as w=μ+3σ−2l bp, where μ and σ are the mean and the SD estimated from mapped read pairs, respectively, and l is the average read length. The inversions detected by BreakDancer were again filtered if they were not detected by the Manta software (Illumina).
In addition, we filtered clinically tolerable inversions observed in the case subject by comparing with inversions observed in normal subjects of Korean ancestry. Inversions of the case subject whose boundaries resided within the window interval w from those of normal subjects were excluded in further analysis.
Fluorescence in situ hybridisation
In order to confirm involvement of the PTH2R gene in chromosomal rearrangement, fluorescence in situ hybridisation (FISH) analysis was performed according to the manufacturer's instructions with commercially available PTH2R FISH probes and the control probe for chromosome 2q11 (Empire Genomics, New York, USA).
Variant calling
After mapping sequence reads to the human reference genome, we marked duplicate sequence reads using the Picard tool and recalibrated base quality scores using the Genome Analysis Toolkit (GATK) V.1.2 TableRecalibration. We realigned sequence reads using the GATK IndelRealigner. We performed variant calling and variant filtering using the GATK UnifiedGenotyper and VariantRecalibrator, respectively. We annotated the variants called with genotype quality >30 using the SnpEff v2.1b software with the GRCh37.66 reference genome and analysed ‘high impact’ variants annotated as nonsense, splice site and frameshift. We excluded variants with minor allele frequency >0.5% in all populations and East populations from the 1000 Genomes and ExAC data (Exome Aggregation Consortium, http://exac.broadinstitute.org (April 2015 accessed)).
Results
The mBAND analysis was carried out and the breakpoints were refined to 2q14 and 2q34 (figure 1B). Array CGH analysis did not reveal any copy number variation in the vicinity of the breakpoints.
To precisely detect the breakpoints for the balanced inversions observed in karyotype analysis, we sequenced the patient's whole genome and generated paired-end sequences of 585 million reads with 118 billion bp. The sequence reads were mapped to the human reference genome and anomalously mapped reads were used to detect structural variants. Among a total of 800 inversions detected from the whole genome, 175 inversions were considered highly confident because these were supported by at least five sequence reads and had a BreakDancer confidence score >80, which was previously shown to indicate ∼90% of the validation rate (see online supplementary table S1 and figure S1) and were not observed in 22 normal Korean individuals.10 Furthermore, 73 inversions were also detected by Manta, and 39 inversions led to chromosomal breaks within 15 kilobases (kb) of nearby genes and, of those, 31 were expected to affect gene function because most regulatory elements fall close to transcribed region (figure 1C and see online supplementary table S2).11 Assuming that the resolution of cytogenetic analysis is about 5 megabases (Mb), we discarded the inversions >5 Mb, which were not detected cytogenetically and were likely false-positives (see online supplementary tables S1 and S2). Analysing all chromosomes left only two inversions involving PTH2R or LRP1B (see online supplementary table S2; lines shaded in light blue) on chromosome 2 to be considered as a gene that might be disrupted to cause the disease. Since both involve 2p14.3 on the left side, only one of the two should be a plausible candidate. However, although the inversion involving LRP1B has a fragment size over 5 Mb, it was not visible in the mBAND analysis which allows delineation of chromosomal regions with a resolution of a few megabases and regarded as a false-positive. The quality per base of sequence reads decreased while the length of the reads increased and the 10th percentile of average quality score at the 60th bp position was lower than 20 (see online supplementary figure S2). To check sensitivity of our analysis, we performed additional analyses by removing the last 41 bp sequences from each read and mapping the trimmed reads to the GRCh37 and Korean (SJK) human reference genomes. All of the eight candidate inversions were detected by analysis of trimmed reads mapped to the two different genomes (see online supplementary figure S3).
Consequently, the disruption of the PTH2R gene was confirmed by FISH analysis (figure 1D). The directly labelled probes for PTH2R (red) and the control locus at 2q11 (green) were used and generated two green and three red (one normal red signal and two split red signals) signals corresponding to the 2q11 locus and PTH2R, respectively. The two small red signals indicated the break of the PTH2R gene, and their close proximity showed that the gene was separated due to an inversion within the chromosome.
To examine if homozygotes of rare variants affect the patient in a recessive form, we analysed variants that were predicted to have high impact on the corresponding protein (nonsense, splice site and frameshift) and were rare (minor allele frequency <0.5% in both all populations and East Asian populations). The patient harboured seven homozygotes for the variants, but none of the corresponding genes was biologically relevant to the disease phenotype (see online supplementary table S3).
Discussion
We used whole genome paired-end sequencing to characterise breakpoints of a balanced chromosomal rearrangement in a patient with non-syndromic midline CRS. The whole genome analysis refined the breakpoint of the inversion to genomic location at 126 101 173 (2q14) and 209 310 827 (2q34) causing separation of the PTH2R gene at intron 7. The 2q14 breakpoint is about 430 kb and 1 Mb away from the nearest transcriptional units (see online supplementary figure S4). Consequently, the PTH2R gene is thought to be fused with the intergenic sequence between CNTNAP5 and GYPC. In genetic diseases, among the possible molecular consequences of chromosome aberrations in genetic diseases,12 it is most likely that the phenotype was the result of deregulation of gene expression through direct disruption of the PTH2R gene in this case.
The PTH2R gene is a member of the family B (type II) of G-protein coupled receptors and is activated by parathyroid hormone and tuberoinfundibular peptide of 39 residues (TIP39) but not by parathyroid hormone-related protein (PTHrP).13 In situ hybridisation revealed that PTH2R is distributed within the cells residing primarily in chondrocytes in the growth plate subarticular cartilage, and the expression was particularly strong in developing bones.14 It was found that both TIP39, a ligand of PTH2R, and PTH2R are expressed in the growth plate of cartilage, with PTH2R being produced by chondrocytes in the resting/early proliferating zone while TIP39 is synthesised by chondrocytes in the prehypertrophic and hypertrophic zones.15 While the main function of PTHrP/PTH1R signalling is to maintain chondrocytes in a proliferative state and prevent hypertrophy, TIP39/PTH2R signalling inhibits the proliferation of chondrocytes.15 In a subsequent study, it was found that targeted expression of PTH2R in growth plate chondrocytes impaired endochondral ossification by decreasing proliferation while impairing differentiation of chondrocytes, which suggests that TIP39/PTH2R has a crucial role in the regulation of chondrocytic proliferation and differentiation.16 Thus, it is likely that disruption of the PTH2R gene in our patient may have caused reduced expression of PTH2R or production of aberrant PTH2R, which led to decreased TIP29/PTH2R signalling and uncontrolled proliferation and differentiation of chondrocytes that in turn resulted in premature closure of sutures.
The IHH protein, which is expressed in prehypertrophic chondrocytes, is known to play a major role in endochondral bone formation by regulating proliferation and differentiation via the PTHrP controlled feedback loop.16 The duplication of locus 2q35, which includes the IHH gene, causes increased IHH expression, which is likely to cause osteoblast proliferation and consecutive overgrowth that results in CRS.17 IHH has also been shown to stimulate chondrocyte proliferation independently of PTHrP signalling, indicating a possible interaction between PTH2R and IHH in regulating chondrocyte proliferation.18 In view of our findings, it will be interesting to determine the function of the PTH2R/PTH2 complex using an animal model of CRS.
Acknowledgments
Whole genome sequence data of Korean control subjects was provided by the Genome Research Foundation, Personal Genomics Institute, and Korean Personal Genome Project.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online figures
- Data supplement 2 - Online tables
Footnotes
JK and H-HW contributed equally.
Contributors JK and H-HW performed experiments, analysed whole genome sequence data and wrote the manuscript. JRC was in charge of the patients, performed mBAND analysis and analysed data. YK and NY performed chromosome analysis, FISH and array CGH. K-AL coordinated and designed the study and reviewed the manuscript.
Funding This study was supported by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A120030).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Institutional Review Board of the Gangnam Severance Hospital (IRB#3-2014-0075).
Provenance and peer review Not commissioned; externally peer reviewed.