Article Text

Abstract
Primary ciliary dyskinesia (PCD) is a rare genetically heterogeneous disorder caused by the abnormal structure and/or function of motile cilia. The PCD diagnosis is challenging and requires a well-described clinical phenotype combined with the identification of abnormalities in ciliary ultrastructure and/or beating pattern as well as the recognition of genetic cause of the disease. Regarding the pace of identification of PCD-related genes, a rapid acceleration during the last 2–3 years is notable. This is the result of new technologies, such as whole-exome sequencing, that have been recently applied in genetic research. To date, PCD-causative mutations in 29 genes are known and the number of causative genes is bound to rise. Even though the genetic causes of approximately one-third of PCD cases still remain to be found, the current knowledge can already be used to create new, accurate genetic tests for PCD that can accelerate the correct diagnosis and reduce the proportion of unexplained cases. This review aims to present the latest data on the relations between ciliary structure aberrations and their genetic basis.
- Cell biology
- Diagnostics
- Genetics
- Molecular genetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Ciliary structure and primary ciliary dyskinesia
Cilia and flagella are highly evolutionary conserved organelles present on the variety of eukaryotic cells. Axoneme, the core of the cytoskeletal structure of cilia and flagella, emanates from the basal body, which is necessary for anchoring the cilium to the apical side of the cell.1 Cilia and flagella are continuously assembled and disassembled at their distal tips. Intraflagellar transport is essential for the transport of flagellar/ciliary components from the basal body to their assembly region and for the removal of turnover products from the flagellar/ciliary tip. Anterograde (base-to-tip) transport with the heterotrimeric kinesin-2 and retrograde (tip-to-base) transport with the cytoplasmic dynein occur at a constant rate without pauses.2–4
The axoneme consists of microtubule (MT) doublets (figure 1) associated with the large number of multiprotein components. In humans, axonemes with either 9+2 or 9+0 microtubular pattern identified on cross sections are present. In the 9+2 cilia, nine peripheral MT doublets surround a central pair (CP) of MTs, while in the 9+0 cilia no CP is present. Cilia can be divided into two major groups: motile and non-motile (primary). Subgroups with different ultrastructural patterns can be distinguished depending on the structure and function they perform:
motile cilia with the 9+2 pattern (nine peripheral microtubular doublets, CP and associated structures like outer dynein arms (ODAs), inner dynein arms (IDAs), nexin-dynein regulatory complexes (N-DRCs), radial spokes (RSs)); found on the apical surface of epithelial cells in the airways (respiratory cilia), brain (ependymal cilia), female reproductive system (cilia in fallopian tube) or in male reproductive system (sperm flagella);5–8
motile cilia with the 9+0 pattern (they lack CP but still contain ODAs and IDAs); found in the embryo (nodal cilia);9 ,10
non-motile cilia with the 9+2 pattern; found in the inner ear (kinocilia);11
non-motile cilia with the 9+0 pattern (no CP and associated structures), acting mainly as mechanosensor and chemosensors; they are present in kidney (renal cilia),12 ,13 bile duct (cholangiocyte cilia),14 pancreas (cilia in pancreatic duct),15 bone or cartilage (cilia in osteocyte or chondrocyte)16 ,17 or in the eye (photoreceptor connecting cilia).18

Schematic diagram of the ciliary axoneme in Chlamydomonas. Upper left: longitudinal section of a cilium presenting the location of axonemal microtubules in different parts of the cilium. Red line indicates the site of a cross section presented on the right site. The cross section shows nine microtubule doublets surrounding the central pair of microtubules. The microtubules are interconnected via radial spokes, N-DRCs and dynein arms; no dynein arms are present in the transition region. All these structures except ODA are distributed with the 96 nm periodicity along the axoneme; ODA are present every 24 nm. The placement of all these structures in relation to each other within the 96 nm repeat is depicted at the bottom. a/d, b/g, c and e, single-headed IDA isoforms; fα and fβ, double-headed IDA isoform f; IC/LC, intermediate chain-light chain complex of inner dynein arms (IDA); Mt A, microtubule A; Mt B, microtubule B; N-DRC, nexin-dynein regulatory complex; ODA, outer dynein arms with marked α, β and γ heavy chains; RS1, radial spoke 1, RS2; radial spoke 2; RS3S, radial spoke 3 stump. Scheme based on the original data published previously.98
The ultrastructural elements of cilia are composed of many proteins; their definition in human cilia is largely based on the information from Chlamydomonas reinhardtii, a unicellular biflagellate green alga.19 A broad knowledge on the function of flagellar proteins could be used in the studies of ciliary proteins thanks to the high evolutionary conservation of these organelles.
Each peripheral doublet consists of MT A and MT B, built of 13 and 11 protofilaments, respectively; protofilaments are composed of α and β tubulin heterodimers. The MTs A and B of the neighbouring peripheral doublets interconnect with each other by the N-DRC. From each MT A, ODAs and IDAs extend; both are large force-producing multiprotein complexes (molecular motors).20 ODA complexes face towards the boundary of the cilium while IDA complexes are directed towards the CP. The peripheral doublets are connected to the CP via RSs. The CP complex, RSs, IDA isoforms f (figure 1) and N-DRC are important for the coordination of the dynein motor activity.21–23 All these components, except ODA, are distributed with the 96 nm periodicity along the axoneme. ODAs are repeated every 24 nm; in addition, two forms of ODA can be distinguished: proximal (characterised by the presence of two heavy dynein chains, DNAH5 and DNAH9) and distal, which do not contain DNAH9.24
Clinical features and diagnosis of PCD
A wide range of different functions played by cilia present on nearly all types of vertebrate cells underlies their importance in the functioning of the organism. Symptoms triggered by dysfunctional cilia form a broad and phenotypically heterogeneous category of overlapping disorders, commonly named ciliopathies.
Primary ciliary dyskinesia (PCD, OMIM id: 242650), a rare, genetically heterogeneous autosomal-recessive disease, is the major ciliopathy related to the dysfunction of motile cilia. The incidence of PCD is estimated at 1:10 000–1:20 000 live births.25 ,26 Extrapolating these data, approximately 70 new cases can be born each year in the white population worldwide.27
The history of PCD dates back to 1904 when Dr A. K. Siewert reported the coexistence of bronchiectasis and situs inversus.28 A few decades later, in 1933, Manes Kartagener described multiple cases with bronchiectasis, situs inversus and sinusitis and named this disorder the Kartagener syndrome.29 Then in 1976, Björn Afzelius, using transmission electron microscopy (TEM), identified an absence of dynein arms in the axoneme of respiratory cilia and sperm flagella in patients with Kartagener syndrome. He concluded that the observed lack of dynein arms must be the cause of ciliary immotility leading to all symptoms of Kartagener syndrome including male infertility due to azoospermia.30
Since then, much more has been learned about the pathophysiology of PCD. This complex disease appears early in life and, if misdiagnosed, may lead to severe symptoms like bronchiectasis or chronic lung disease.31 ,32 PCD symptoms involve organs where motility of cilia has an impact on their normal functioning. The most prominent PCD features relate to the upper and lower respiratory tract and usually appear early after birth. Those symptoms include neonatal respiratory distress syndrome, which comprises chest congestion, coughing, tachypnoea (rapid breathing) and hypoxia.31 ,32 In cases where dextrocardia, situs ambiguous or situs inversus are present in the infant, PCD should be considered as a highly possible diagnosis.33 ,34 Later, chronic sinusitis and secretory otitis media might occur, which, together with chronic middle ear effusion, frequently lead to hearing loss.35–37 There are also lower respiratory tract symptoms like wheezing, chronic wet cough frequently with sputum production, chronic bronchitis and recurrent pneumonia; after several years these symptoms may lead to bronchiectasis in the middle and lower lobes.27 Upper respiratory tract abnormalities include persistent rhinitis, mucosal congestion, nasal passages oedema and infrequently (more prevalent in adults) nasal polyps.38 In older children and adults, chronic and recurrent sinusitis as well as chronic mucopurulent sputum production are common features. Bacteria commonly identified in sputum samples after microbiological testing in those patients are Haemophilus influenzae, Staphylococcus aureus and Streptococcus pneumoniae.39 Older individuals with advanced lung disease exhibit greater frequency of Pseudomonas aeruginosa and non-tuberculous mycobacteria occurrence.31 ,38 With time, lung functions may deteriorate to the stage of a severe respiratory failure, when lobectomy and lung transplantation are highly recommended.26 Moreover, male infertility due to the sperm tail dysmotility, as well as decreased fertility in women, is observed.31
Recognition of PCD and the correct diagnosis are often delayed due to the clinical symptoms overlapping with other chronic airway disorders. The diagnosis is easier when the patient exhibits abnormal placement of the internal organs observed on the chest X-ray.25 ,26 As the disease is progressive, late recognition may result in a worse prognosis for patients due to an inadequate previous treatment. Thus, PCD diagnosis requires a well-described clinical phenotype combined with the identification of abnormalities in the ciliary ultrastructure and/or beating pattern.
Patients with PCD exhibit distinct structural and functional defects of cilia, ranging from almost normal ultrastructure but abnormal beat pattern, through the lack of dynein arms resulting in cilia immotility, to a complete absence of cilia.24 ,40–44 Up to now, a combination of several diagnostic methods/techniques is used to properly identify the disease, including nasal nitric oxide measurement,45 TEM analysis of the ciliary ultrastructure,46–48 high-resolution immunofluorescence (IF) microscopy49 ,50 and high-speed video microscopy (HSVM) analysis of ciliary waveform and the beat frequency.32 In some cases, to differentiate PCD from clinically similar secondary ciliary dyskinesia, ciliogenesis de novo is required, and cultures of the respiratory epithelium cells (submerged51 or air–liquid interface38 ,52) are performed.
Correct recognition of PCD can be aided by performing genetic tests, which would detect underlying genetic causes of the ciliary dysfunction. However, because of the complexity of the ciliary structure and function, PCD is genetically highly heterogeneous. Recently, the availability of high-throughput sequencing techniques contributed to the swift identification of new PCD-causative genes (further referred to as PCD genes), but with almost 30 such genes identified so far, genetic causes of approximately one-third of PCD cases still need to be explained.
Genetics of PCD
The identification of PCD genes has been based on linkage studies, candidate gene approaches and proteomic analyses, combined with sequencing of potentially causative genes.53 ,54 Linkage analysis and homozygosity mapping have been used to indicate the regions of interest in the genome of PCD families and cohorts.54 ,55 Selection of the candidate genes was based on data from model organisms; in particular, Chlamydomonas mutants with the defective structure and/or function of flagella delivered many candidate genes for PCD studies in human.53 ,56 ,57 For many years, search for mutation in the candidate genes was based on Sanger sequencing. Recently, the next-generation whole-exome and whole-genome sequencing technology became available, revolutionising identification of new mutations and new genes related to PCD.
Table 1 provides a comprehensive list of PCD-related genes. To date, mutations in 27 genes have been identified in patients with classical PCD symptoms. Two more genes, RPGR and OFD1, are mutated in rare syndromic PCD cases (combined with retinitis pigmentosa and orofacialdigital syndrome, respectively).58–60 Mutations in another gene, CCNO, found in 16 patients, are involved in the disease characterised by the paucity of motile cilia, with the PCD-like clinical consequences. CCNO gene encodes cyclin O, and its mutations result in the defective generation and placement of the mother centriole.44
A comprehensive list of primary ciliary dyskinesia (PCD)-related genes
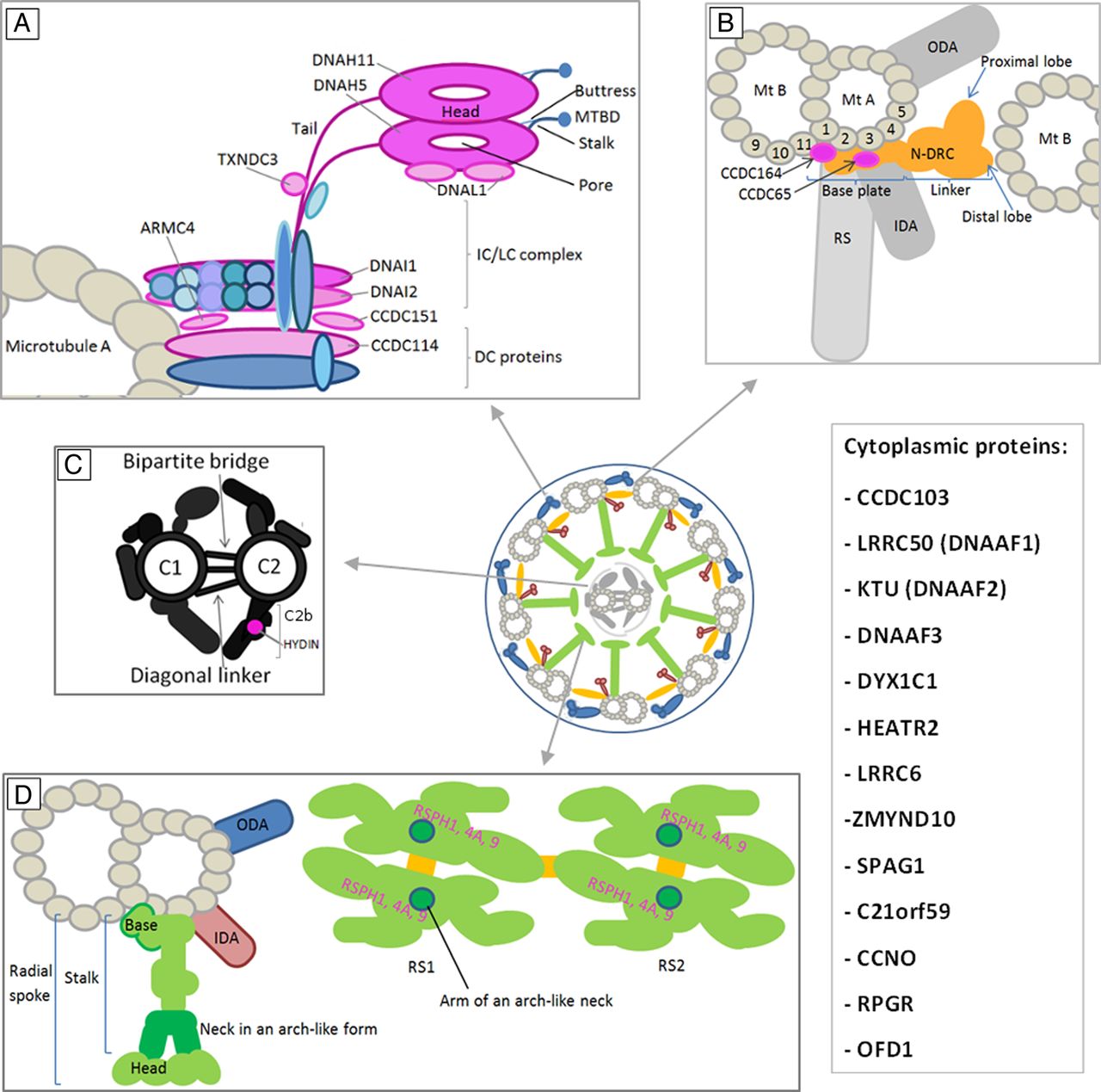
The PCD genes can be divided according to their protein product location and function, or to the ultrastructural/functional defect in patients with PCD. These aspects are summarised in table 1 and figure 2. Figure 2 provides a schematic representation of the probable localisation of PCD-relevant proteins within the human ciliary structure components.

{kind=link}
{kind=link}
The probable localisation of identified primary ciliary dyskinesia (PCD) genes products within the human ciliary structure components. The schemes of human axonemal complexes are adapted from Chlamydomonas data. The proteins related to PCD are indicated in purple and their names are provided. Cytoplasmic proteins involved in PCD phenotype are listed in the box on the right. (A) ODA consists of heavy dynein chains β and γ (comprising a tail, the AAA-ring-like structure, which is a motor unit, and the stalk with MTBD and coiled-coil protrusion called strut or buttress, which strengthens the base of the stalk), LCs, ICs and DC proteins. (B) N-DRC consists of the base plate and linker that further comprises proximal and distal lobes. (C) CP scheme involves microtubules C1 and C2 with their projections (including C2b) interconnected by a bipartite bridge and diagonal linker. (D) RS structure is depicted in a cross-sectional (left) and a bottom (right) view. RSPH1, 4A and 9 are components of RS head (pink). CP, central pair; DC, docking complex; IC, intermediate chain; IDA, inner dynein arm; LC, light chain; Mt A and B, microtubule A and B; MTBD, microtubule binding domain; N-DRC, nexin-dynein regulatory complex; ODA, outer dynein arm; RS, radial spoke; β and γ, dynein heavy chains β and γ. Schemes based on the original data published previously.20 ,21 ,87 ,99–101
Genes encoding ODA components
Mutations in these genes result in a typical defect of the axonemal ultrastructure, characterised by the absence or severe shortening of ODA. This can be observed in TEM cross sections (lack of ODA) or in IF analysis using antibodies directed against the ODA marker proteins (no reaction with anti-DNAH5 and anti-DNAH9).50 The defects cause complete or almost complete immotility of cilia.61
The first PCD gene, DNAI1, identified in 1999 using a candidate gene approach,53 is the human ortholog of Chlamydomonas IC78 gene and encodes intermediate dynein chain 1 of ODA. The majority of DNAI1 mutations, clustering in three exons (13, 16 and 17) and in intron 1 (IVC1+2_3insT), explain up to 14% of PCD cases.62–65
The second identified gene, DNAH5, discovered using homozygosity mapping and linkage analysis performed in one consanguineous family,54 is the human ortholog of Chlamydomonas gene encoding γ-axonemal heavy dynein chain. DNAH5 encodes heavy dynein chain 5, the major molecular motor of ODAs. Mutations in DNAH5 identified in further studies42 ,43 account together for more than 25% of all PCD cases.43 The most frequently found mutations cluster in exons 34, 50, 63, 76 and 77.49
Other genes encoding structural components of ODA, DNAI2, DNAL1 and TXNDC3, are less frequently involved in PCD. Mutations in DNAI2, encoding intermediate dynein chain 2, were identified using candidate gene approach24; mutations have been found in five PCD families so far.7 ,24
TXNDC3 (also known as NME8) was identified as the human ortholog of the sea urchin ODA component IC166; two compound mutations have been found in a single patient. DNAL1 was considered a good candidate for PCD since DNAL1 protein was shown to specifically bind DNAH5. But for a long time the search for mutations was ineffective.65 Finally, a homozygous point mutation in DNAL1 was found in two Bedouin patients with absent or shortened ODAs.67
IF analysis of the respiratory cilia from PCD individuals with DNAI2, DNAH5 and DNAI1 mutations revealed that mutations in DNAI1 affect assembly of proximal ODA complexes (containing DNAH5 and DNAH9), while mutations in DNAI2 and DNAH5 disturb the assembly of both ODA complexes, proximal and distal (which do not contain DNAH9).24
Genes related to assembling of dynein arms
Multisubunit dynein arm complexes, produced and preassembled in the cytoplasm, are transported to the cilia and anchored into the axonemal MTs via the ODA docking complex. Recently, mutations in a number of genes involved in the assembly, targeting and docking processes have been identified in individuals with ODA or combined ODA/IDA defects.
ODA defects have been shown to result from mutations in CCDC114, encoding ODA-docking complex component required for attachment of ODA to the MT A,68 and in CCDC151, encoding protein required for the axonemal assembly of ODA-docking complexes.69 Mutations in these two genes have been found in singular PCD families. More frequent mutations have been found in ARMC4, encoding the protein, which may be important in targeting or anchoring of ODA to the MT A.70 ,71
Furthermore, cytoplasmic proteins were identified, which most probably play a role in preassembly or transport of both ODA and IDA components. KTU (DNAAF2), encoding protein localising to the apical part of the cell cytoplasm, was the first preassembly factor described to cause PCD.75 It was identified in medaka fish mutant with dynein arms defect; the loss of both dynein arms and immotility of cilia have been observed in two patients with mutations in KTU.75 Other genes encoding factors involved in the cytoplasmic preassembly pathway in PCD pathogenesis are: rarely involved LRRC50 (DNAAF1),55 ,56 DNAAF3,76 HEATR278 and C21orf59;83 frequently or very frequently involved DYX1C1 (protein interacts with KTU),77 LRRC6,7 ,79 ,80 ZMYND10 (protein interacts with LRRC6)7 ,81 and SPAG1.82 Mutations in all these genes lead to the combined ODA and IDA deficiency.
IF analysis in individuals (mostly Pakistani) with mutations in CCDC103 demonstrated that respiratory cilia partially lack ODA complexes (with the ODA marker protein DNAH5 accumulating in the apical part of cytoplasm) but exhibit normal localisation of IDA marker protein DNALI1.74 CCDC103 has been proposed to act during ODA assembly in the cytoplasm as well as in the proximal part of cilia.74
Genes encoding radial spokes or CP component
Abnormalities in MTs arrangement involving absence of the CP, with the 9+0 pattern or 8+1 pattern (one of peripheral doublets shifted towards the axoneme centre), are the most common feature observed in TEM cross sections in patients with mutations in some genes.31 ,93 These defects result in a range of altered beat patterns.61 ,87
RSPH4A and RSPH9 are radial spoke head proteins and most probably play a role in signal transduction between CP and dynein arms as they interact with CP on their head side and with IDA on the other end (figures 1 and 2). Mutations in RSPH9 and RSPH4A have been first detected using linkage analysis and homozygosity mapping in several consanguineous Bedouin families that shared PCD phenotype with CP–RS abnormalities.87 Other mutations in RSPH4A are relatively frequent in other populations.88 ,89 ,94 In the East European PCD population study, no RSPH9 mutations were found, suggesting they could be exclusively related to Bedouin families88; recently, other RSPH9 mutations have been reported in four families of unspecified origin.89 Recently, mutations in another gene encoding radial spoke head protein, RSPH1, have been detected in a number of patients with PCD with the CP–RS defect89–91; RSPH1 appears to be the most common gene mutated in individuals with this particular defect.89
Mutations in HYDIN, found so far in five families, cause much more subtle defects.92 Mutant cilia lack the C2b projection in the CP complex (figure 2), but most TEM cross sections appear normal, with no MT disorganisation typical for mutations in the radial spoke proteins. Patients demonstrate only subtle abnormalities in the ciliary waveform, which can be correctly recognised only when aided by the high-speed videomicroscopy analysis.92 These features impede correct diagnosis of PCD in patients with HYDIN mutations.
Genes encoding proteins of the N-DRC
The studies on Chlamydomonas mutants provided an insight into the complexity of the N-DRC structure, and a number of protein components of N-DRC have been identified, including DRC4, DRC1 and FAP250.22 ,83 ,86 IF analysis of human cilia performed using GAS11 (aka GAS8), the human ortholog of DRC4, has been used as a protein marker of N-DRC in IF studies in human PCD.50 So far, no mutation has been identified in GAS11 but mutations in other N-DRC proteins have been found.
The first two mutant genes associated with defects in N-DRC and IDA complexes in PCD were CCDC39 and CCDC40.57 ,84 ,85 Broad analysis of the axonemal structure and function in the individuals with mutations revealed the same defects. The cilia lacked N-DRC, IDA and RS, which resulted in MT disorganisation, and the hyperkinetic and stiff ciliary beat pattern.57 ,84 ,85 Such a wide range of ultrastructural defects indicated that both proteins could be components of another axonemal complex related or attached to N-DRC.86 ,87 Mutations in these genes have been found in a large number of patients with PCD.
Two other genes related to N-DRC defects were found in a few families. Patients with PCD with mutations in CCDC164, a human ortholog of Chlamydomonas DRC1, exhibited the absence of N-DRC but no IDA or MT defects were found.86 Moreover, it has been shown that CCDC164 mutations do not disturb proper localisation of CCDC39. The impairment of the ciliary beat pattern and beat frequency observed in CCDC164 mutant cilia were less severe than in CCDC39 mutants.86
Patients with mutations in CCDC65, the human ortholog of Chlamydomonas FAP250, displayed reduction of IDA and N-DRC, and stiff ciliary beat pattern.83 Moreover, some TEM cross sections showed MT disorganisation advocating the role of N-DRC in stabilisation of axonemal structures organisation.83
Genes responsible for abnormalities in ciliary movement with normal ultrastructure
Some patients with PCD presenting clinical manifestations consistent with PCD phenotype have no detectable ultrastructural defects of cilia. In these cases, however, abnormalities in the ciliary beat pattern can be found. Cilia are stiff (nonflexible) and hyperkinetic40 or static,95 resulting in the impaired mucociliary clearance and clinical PCD phenotype. So far, DNAH11, encoding heavy dynein chain 11 of ODA, has been the only identified gene related to such a phenotype.40 Mutations in this gene are relatively common in PCD individuals with normal axonemal ultrastructure and explain about 20% of this particular phenotype.41
Genetic testing in PCD
For a long time, genetic testing available was based on the identification of the most common mutations in two genes responsible for the ODA defect: IVS1+2_3insT and mutations in exons 13, 16 and 17 for DNAI1, and exons 34, 50, 63, 76 and 77 for DNAH5.49 Mutations in these two genes explain about 50%–60% of PCD cases with the ODA defect. With ODA defects observed in ∼60% of PCD cases, mutations in DNAH5 and DNAI1 can be expected in only 30%–35% of the whole PCD population.49 This proportion well illustrates the fact that genetic testing comprising only selected genes in a heterogeneous disease like PCD is not sufficiently effective. Moreover, genotyping only the most frequent mutations in genes characterised by high allelic heterogeneity often results in the identification of only a single mutated allele, which is not sufficient for the diagnosis; in such cases, the whole gene should be screened to search for the second, trans-allelic mutation.49 The scope of mutations has been expanded including other PCD genes in addition to the first two ones.96
The one-by-one screening of a number of genes in genetically heterogeneous disorders like PCD is, therefore, neither time-effective nor cost-effective. The whole-exome sequencing (WES) enables rapid and relatively reasonably priced identification of mutations in genetically heterogeneous disorders like PCD. The WES approach has been recently used for the identification of new genes associated with PCD,7 ,68 ,70 but this technology is still reserved for the research field and not applied in the diagnostics. However, with dropping sequencing costs, there is a chance for WES to become a routine test in the future. Indeed, this is currently happening: a broad diagnostic gene panel service has just been initiated within the North Thames Regional Genetics service provided at Great Ormond Street Hospital.97
Concluding remarks and future perspectives
A rapid acceleration in the pace of identification of PCD-causing genes during the last 2–3 years is notable. This is the result of new technologies that have been launched recently and applied in genetic research, like whole-genome SNP-based homozygosity mapping or WES. Nevertheless, genetic causes of approximately one-third of PCD cases remain to be explained.
The genes already identified can be used in genetic screening, enabling earlier recognition of PCD and implementation of an appropriate treatment from the earliest age. This would have a huge impact on the patients, vastly improving the quality of life, professional and private. In this context, it is important that many of the PCD genes identified so far are related to particular PCD phenotypes, which facilitates and simplifies genetic screening if preceded by TEM, IF or HSVM analysis.
It should be emphasised that while early studies focused on the identification of genes encoding proteins that build easily recognised ultrastructural components of cilia, like ODA, IDA or RS, the recent advances in understanding of the ciliary structure and assembly have shifted the attention to the genes encoding proteins involved in the processes of assembling, targeting and docking of these ciliary components.
The recent findings indicate the existence of sequential stages of dynein arms assembly. Functional characterisation of the genes encoding cytoplasmic proteins, which are not a part of the axoneme structure, can help to explain the processes during the axoneme assembly pathway. Moreover, cytoplasmic proteins involved in ciliogenesis might be more sensitive/responsive to potential drugs and new treatment modalities due to their better physical accessibility than the structural axonemal proteins.
Acknowledgments
MK is supported by the project 'Studies of nucleic acids and proteins—from basic to applied research', sponsored by the International PhD Projects Programme of Foundation for Polish Science. The project is co-financed by the European Union Regional Development Fund. MK is also supported by the project 'Scholarship support for PhD. students specialising in majors strategic for Wielkopolska's development', Sub-measure 8.2.2 Human Capital Operational Programme, co-financed by European Union under the European Social Fund. EZ is supported by the National Science Centre (grant no. 2011/01/B/NZ4/04840). This work has been supported from the European Union Seventh Framework Programme (FP7/2007-2013) funds under grant agreement no. (305404) (BESTCILIA) (MW).
References
Footnotes
Contributors MK performed the literature research, prepared figures and table, and wrote the manuscript. EZ and MW reviewed the paper.
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.