Article Text

Abstract
Background The majority of human embryos created using in vitro fertilisation (IVF) techniques are aneuploid. Comprehensive chromosome screening methods, applicable to single cells biopsied from preimplantation embryos, allow reliable identification and transfer of euploid embryos. Recently, randomised trials using such methods have indicated that aneuploidy screening improves IVF success rates. However, the high cost of testing has restricted the availability of this potentially beneficial strategy. This study aimed to harness next-generation sequencing (NGS) technology, with the intention of lowering the costs of preimplantation aneuploidy screening.
Methods Embryo biopsy, whole genome amplification and semiconductor sequencing.
Results A rapid (<15 h) NGS protocol was developed, with consumable cost only two-thirds that of the most widely used method for embryo aneuploidy detection. Validation involved blinded analysis of 54 cells from cell lines or biopsies from human embryos. Sensitivity and specificity were 100%. The method was applied clinically, assisting in the selection of euploid embryos in two IVF cycles, producing healthy children in both cases. The NGS approach was also able to reveal specified mutations in the nuclear or mitochondrial genomes in parallel with chromosome assessment. Interestingly, elevated mitochondrial DNA content was associated with aneuploidy (p<0.05), a finding suggestive of a link between mitochondria and chromosomal malsegregation.
Conclusions This study demonstrates that NGS provides highly accurate, low-cost diagnosis of aneuploidy in cells from human preimplantation embryos and is rapid enough to allow testing without embryo cryopreservation. The method described also has the potential to shed light on other aspects of embryo genetics of relevance to health and viability.
- Aneuploidy
- Obstetrics and Gynaecology
- Cytogenetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Chromosome segregation during female meiosis is particularly error prone in humans, a problem that worsens with advancing age. Recent studies have demonstrated that approximately a quarter of oocytes from women in their early 30s are chromosomally abnormal, with aneuploidy rates increasing to over 75% in the oocytes of women over 40.1 It has been shown that most of the aneuploid embryos produced from such oocytes fail to implant in the uterus, although a minority do succeed in forming a pregnancy only to later miscarry.2 The high frequency of chromosome abnormality during the first few days of life is of great relevance to infertility treatments such as in vitro fertilisation (IVF). Typically, IVF involves the fertilisation of several oocytes, but in order to avoid multiple pregnancy and the associated risks of complications for the mother and children, it is recommended that the number of embryos transferred to the uterus is restricted, ideally to a single embryo.3 Maximising the likelihood of obtaining a pregnancy depends on accurate determination of the embryo with the greatest capacity for producing a child, ensuring that it is prioritised for transfer. Currently, the decision of which embryo should be transferred is primarily based on a simple evaluation of morphology. However, the appearance of an embryo is only weakly correlated with its potential to form a pregnancy and reveals no useful information about its chromosomal status. In theory, screening for aneuploidy, a problem which has a more definitive impact on the ability of an embryo to produce a healthy baby, could provide a valuable means of identifying viable embryos.
The main obstacle to testing human embryos for aneuploidy is the extremely limited amount of tissue available for analysis. At the time when most genetic testing has traditionally been carried out, embryos are composed of just 6–10 cells and only one cell (blastomere) may be safely removed for analysis. Although obtaining accurate genetic information from a single cell is challenging, recent years have seen several methods developed for this purpose. Some protocols are based on the use of microarrays (eg, comparative genomic hybridisation or analysis of single nucleotide polymorphisms),4 ,5 while others involve quantitative PCR.6 In the last 2 years, several randomised trials using these techniques have been undertaken, producing clinical data supporting the hypothesis that screening of embryos for aneuploidy can improve IVF outcomes, increasing pregnancy rates and reducing miscarriages.7––10 However, all methods currently available for the genetic analysis of preimplantation embryos suffer from shortcomings which limit their clinical applicability.
Cost is an important consideration for both clinical and scientific applications of single cell analysis. In the context of IVF treatment, the cost of aneuploidy screening is multiplied by the number of embryos produced by the couple (averaging approximately six, but in some cases exceeding 20). The expense associated with the testing of multiple samples puts chromosome analysis beyond the reach of many patients. The potential for producing a large number of embryos in a single cycle of IVF treatment also means that any test must be scalable, allowing multiple samples to be assessed simultaneously. For any clinically applied diagnostic assay, it is advantageous if results can be provided rapidly. However, in the case of preimplantation embryo testing the speed of the procedure assumes even greater importance since the window of time during which the embryo has the capacity to implant and the mother's endometrium is receptive is narrow. Depending on the exact embryo biopsy strategy employed, there may be less than 24 h available for genetic analysis.
This study aimed to develop a rapid, scalable, cost-effective method for the genetic analysis of single cells (blastomeres) or trophectoderm biopsies derived from human preimplantation embryos. For this purpose, we chose to employ next-generation sequencing (NGS) technology. NGS is a term that encompasses a variety of different methods capable of generating large quantities of DNA sequence information, rapidly and at a low cost per base. Our data confirm that NGS can be successfully applied to the diagnosis of a variety of genetic abnormalities in single cells from human embryos and has various advantages over traditional technologies used for preimplantation genetic diagnosis and screening. Clinical applicability was demonstrated.
Results
Optimisation of an NGS protocol for use with whole genome amplification products from single cells
Optimisation of the NGS protocol was performed in order to maximise the quantity of data (ie, number of reads) obtained per sequencing run. The number of reads per run is an important consideration, as the greater the amount of sequence data produced the larger the number of samples that can be simultaneously tested. Multiplex analysis of samples is an essential factor in lowering the per-embryo costs of NGS. Efforts were also made to reduce the time from sample acquisition to results, which is important given the extremely limited time available for the assessment of preimplantation embryos. Protocol improvements increased the amount of sequence data obtained more than threefold compared with initial results, producing an average per chip of 356.62 Mb.
A time series of incubations was performed on a range of input DNA concentrations (100–500 ng) of whole genome amplification (WGA) product to establish the optimal fragmentation conditions for samples derived from single cells. An amount of 100 ng of input DNA was found to be sufficient for generating successful sequencing libraries and was used for all subsequent samples in order to minimise the amount of WGA necessary. Reducing the amount of amplification decreases the risk of artefacts and allows for an accelerated protocol. Accurate quantification of the input material was essential for enabling this lower range of WGA DNA to be used. Quantification using a Nanodrop spectrophotomer consistently resulted in an overestimation of the DNA concentration resulting in insufficient input material being used for library generation. The Qubit high sensitivity double stranded DNA (HS dsDNA) quantification method was found to give a much more accurate estimation of the amount of DNA present in the sample. Incubation times of 15 and 17.5 min provided the optimal size fragment distribution for Ion Torrent 100 and 200 bp chemistries, respectively.
Agarose and solid phase reversible immobilisation (SPRI) bead-based methods were both tested for size selection of the sequencing libraries. For the SPRI bead size selection, the AxyPrep FragmentSelect-I kit (Appleton Woods) was trialled. A ratio of 2× input sample volume to 1.8× input volume of binding to wash off was used to select the fragment at 300 bp. This method was reproducible, rapid and effective for fully removing the upper and lower fragments of DNA, but resulted in a relatively broad size range distribution. This negatively affected the amount of data generated on the Ion Torrent PGM sequencer, as the shorter fragments were preferentially amplified. Gel excision was found to give a much tighter distribution of read lengths. The sample fraction at 300 bp was excised using E-Gel SizeSelect Gels (Life Technologies).
Both 100 and 200 bp sequencing chemistry were found to be appropriate for analysis of aneuploidy and DNA sequence in single cells. Ultimately, in order to generate as much data as possible for each sample, the 200 bp chemistry was selected. A minimum of five cycles of adaptor mediated amplification was required to generate quantifiable sequence-ready libraries. The samples were quantified on a 2100 Bioanalyser High Sensitivity Chip (Agilent Technologies, Santa Clara, California, USA). Clonal amplification for template preparation was performed on the Ion OneTouch system. Input concentrations of 18 and 24 pM were tested to establish the optimal sample to bead ratio for template preparation. An amount of 24 pM input concentration resulted in a greater yield of data without generating significantly more polyclonal reads.
Aneuploidy can be detected in single cells from embryos using a rapid low-pass genome sequencing methodology
The current research focused on the use of multiple displacement amplification (MDA) in order to generate sufficient DNA for subsequent NGS. Amplification was obtained from 61/61 (100%) samples. Initially, NGS was applied to single cells isolated from six well characterised, karyotypically stable cell lines (samples 1–18 in table 1).
Samples assessed and cytogenetic results obtained
From the analysis of single cells of known karyotype, it was clear that WGA introduced distortions in the relationship between the number of sequence reads and chromosome length, complicating attempts to assess aneuploidy based on reads per chromosome. However, these deviations were found to be highly reproducible from one sample to the next, presumably a consequence of preferential DNA amplification associated with differences in the base composition and chromatin structure of individual chromosomes. The consistency of the distortions meant that it was relatively simple to compensate for amplification bias. This was achieved by comparing results obtained from cell line and embryo cells with those obtained from a series of chromosomally normal reference samples. Essentially, a set of 24 reference values were created by averaging the percentage of mapped reads attributable to each chromosome in a series of euploid samples (see online supplementary table S1). For embryo (test) samples, the percentage of reads derived from each chromosome was divided by the reference value for the same chromosome. Chromosomes present in a normal (disomic) state displayed a test to reference ratio ranging from 0.7 to 1.2, whereas chromosomal gain (eg, trisomy) and loss (eg, monosomy) were associated with ratios >1.2 and <0.7, respectively. Using this approach, the karyotypes of all single cells derived from cell lines were correctly defined using NGS (summarised in table 1).
NGS provides a highly accurate detection of aneuploidy and confirms that monosomy and other unusual forms of aneuploidy remain common at the final stage of preimplantation development
During the initial validation phase of this study, 38 samples from human embryos were analysed using NGS. Results were obtained from all embryos tested (100%). The samples assessed included cells from 32 blastocysts, the final stage of preimplantation development. The blastocysts were derived from five infertile couples of advanced reproductive age (average female age 42 years). Overall, 24 of the embryos were diagnosed abnormal (75%), emphasising the high risk of transferring an aneuploid embryo to the uterus when such patients undergo IVF treatment. A total of 51 chromosome errors were detected (table 1; see online supplementary figure S1). With the exception of chromosomes 3, 4, 5 and 20, all chromosomes were affected by aneuploidy at least once in this set of samples, with chromosome 22 displaying the greatest number of errors. Monosomies and trisomies occurred at similar frequencies.
A second biopsy was taken from each embryo, coded and blindly assessed using a well-established microarray-comparative genomic hybridisation (CGH) approach. After decoding, the diagnostic results from the two analyses were compared. In all cases, the tests were in agreement concerning the embryos that were chromosomally normal and those that were aneuploid (figure 1). The karyotypes predicted by microarray comparative genomic hybridisation (aCGH) and NGS were entirely identical for all but two embryos. The embryos with discrepant results (numbers 34 and 37 in table 1) were considered highly abnormal using both methods, each affected by several aneuploidies (>3 abnormal chromosomes each). Considering the 1296 chromosomes assessed in the 54 samples used to evaluate the accuracy of NGS, the concordance rate per chromosome was 99.7%.
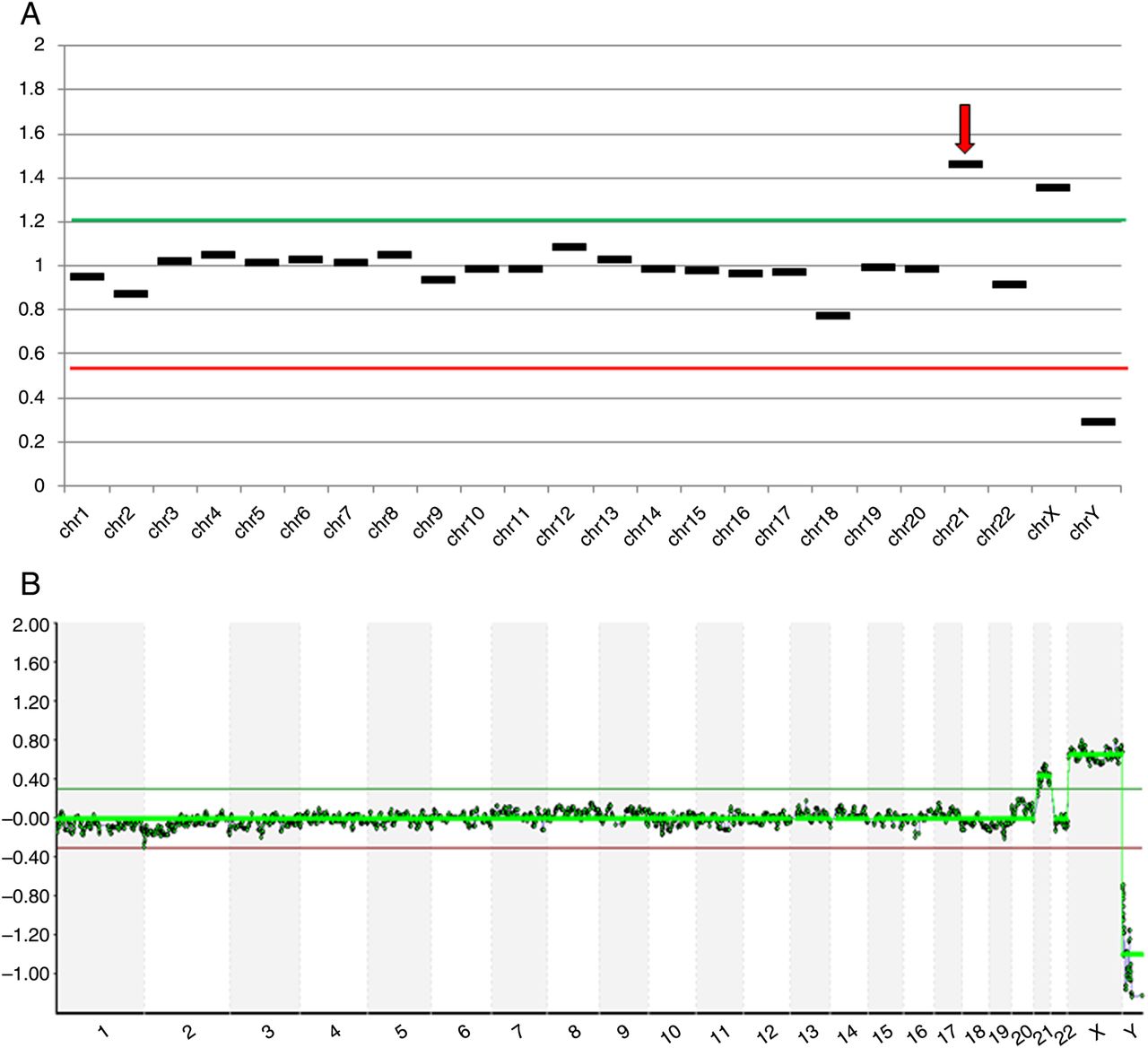
Aneuploidy analysis of cells biopsied from a human blastocyst. (A) Analysis of embryo #41 using next-generation sequencing (NGS) predicting a female trisomic for chromosome 21; (B) Microarray-CGH analysis of a second embryo biopsy sample from embryo #41, confirming the NGS result.
Chromosomal analysis of embryos using NGS can be carried out at a speed, throughput and cost appropriate for use in conjunction with standard embryo biopsy and transfer protocols
Given the quantity of sequence data produced from each sequencing chip, it seemed likely that a large number of samples could be simultaneously tested, significantly reducing costs per sample and greatly increasing throughput. To verify this, 32 samples derived from cells biopsied from human embryos were multiplexed together. Different adapters of unique DNA sequence (ie, ‘barcodes’) were ligated to the DNA fragments obtained from each embryo and samples were pooled together and then sequenced simultaneously. Once the process was complete, the sequence of the barcode at the beginning of each DNA fragment was examined allowing the fragment to be assigned to an individual embryo (see online supplementary figure S2). The cytogenetic diagnoses obtained were found to be identical regardless of whether cells were analysed individually or multiplexed together. The robust results obtained for the simultaneous analysis of 32 samples suggest that even higher levels of multiplexing are likely to be possible.
Chromosomal analysis of cells from embryos could be completed within 15 h using the optimised NGS protocol. However, even faster testing was shown to be possible using the new Ion Isothermal Amplification Chemistry (Life Technologies), which streamlines the sequencing procedure, eliminating the need for emulsion PCR. Using this approach, chromosome screening data could be obtained within 8 h (see online supplementary table S3). A total of 17 embryo biopsy specimens were simultaneously tested using this new approach and chromosomal copy number assessments were shown to be concordant with aCGH in all cases (see online supplementary table S4).
NGS allows the potential for simultaneous chromosomal analysis and diagnosis of gene mutations in single cells
In the current study, low-pass sequencing of WGA products yielded <0.1% coverage of the genome, so the chances of a given locus being represented were extremely low. For this reason, attempts to provide data on specific mutations required a semitargeted approach, enriching the genomic sequences relevant for diagnosis (ie, the mutation sites and/or informative linked polymorphisms) before carrying out NGS. As a proof of concept, single cells were isolated from a cell line homozygous for the common CFTR ▵F508 mutation, associated with cystic fibrosis. After cell lysis and WGA, an aliquot of the amplified product was subjected to PCR, amplifying a 95 bp DNA fragment encompassing the ▵F508 mutation site. Sequencing libraries were generated from the original WGA product and the locus-specific PCR product. The two libraries were then combined together and simultaneously sequenced. This approach provided sufficient DNA sequence information across all chromosomes to allow detection of aneuploidy, while also sequencing the region of CFTR containing ▵F508 multiple times (sequenced to a coverage depth of approximately 30×). All of the CFTR sequence reads obtained confirmed the homozygous-affected genotype of the tested cells (see online supplementary figure S3).
The NGS method provides quantitative data on mtDNA copy number and mutation load
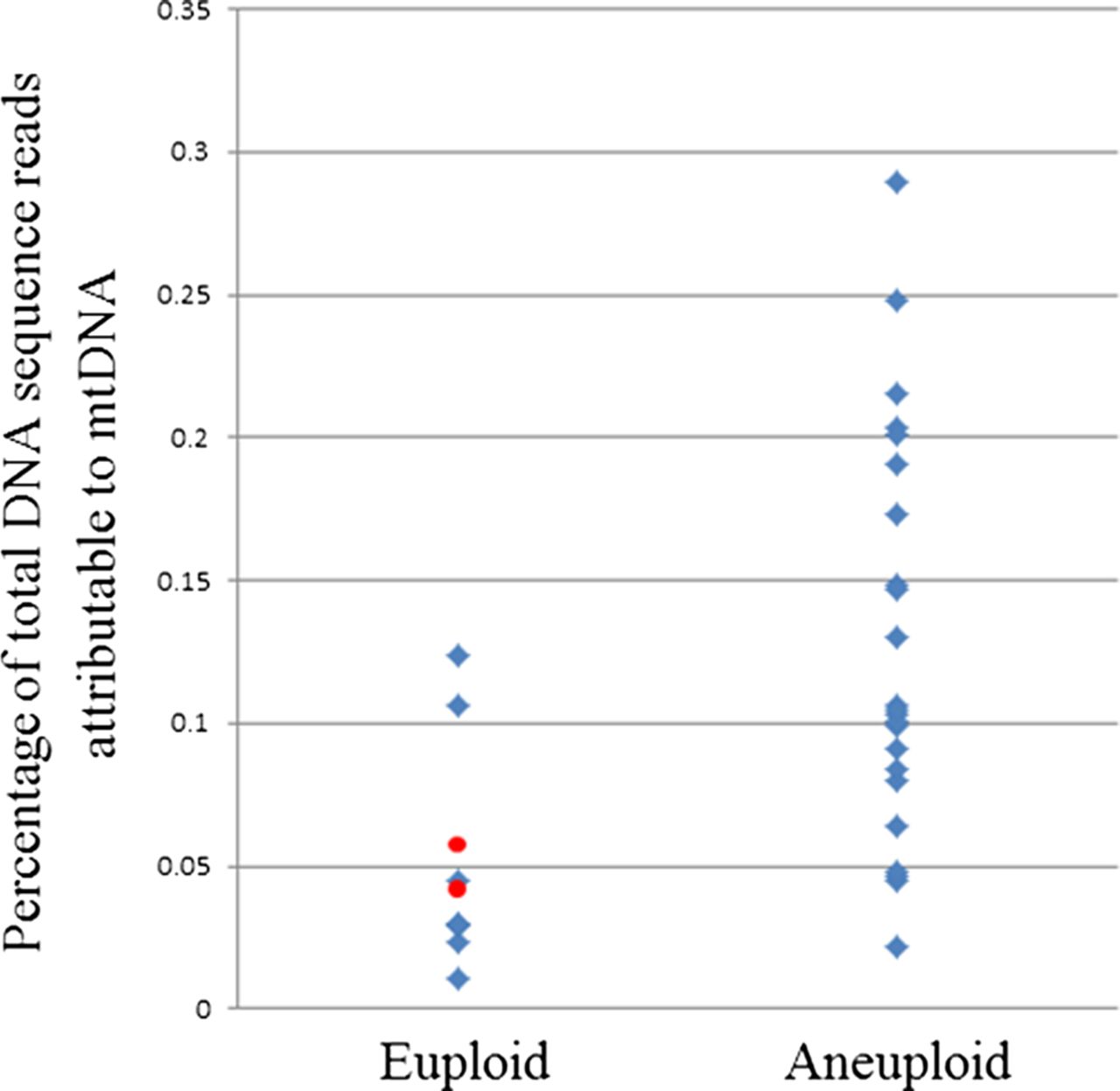
Not only did the optimised NGS protocol allow accurate detection of aneuploidy, it also succeeded in sequencing the entirety of the mitochondrial genome in single cells to a coverage depth of 20–60×. Interestingly, when the ratio of mitochondrial DNA (mtDNA) sequences to nuclear DNA sequences was considered, significant differences were seen between embryos derived from the same couple. In most cases, the embryo with the largest quantity of mtDNA from a single couple had three to sixfold more than the embryo with the least. However, more extreme variations were also detected. In one instance, a 97-fold difference in mtDNA content was observed between two embryos derived from the same parents. It seems highly likely that such dramatic differences in a key organelle would have functional consequences for the affected cells. Indeed, a potentially important observation was that embryos with high levels of mtDNA had an elevated risk of aneuploidy (p<0.05) (figure 2). The use of real-time PCR to quantify a fragment of the mitochondrial genome in trophectoderm samples from human blastocysts verified the results of NGS-based mtDNA quantification (see online supplementary figure S4).

{kind=link}
{kind=link}
Relationship between blastocyst aneuploidy and relative mitochondrial DNA (mtDNA) quantity in human blastocyst stage embryos. The amount of mtDNA relative to nuclear DNA is significantly lower in biopsies of trophectoderm cells from euploid embryos (p<0.05, two-tailed t test). Red spots indicate the mtDNA content of the two chromosomally normal embryos that were transferred to patients and produced viable pregnancies.
In the current study, a blinded analysis was carried out on individual cells isolated from a cell line affected with an mtDNA disorder. Analysis of DNA from the cell line using conventional methods had previously shown that ∼70% of the mitochondrial genomes were affected by a clinically significant 7438 bp deletion. The single cell NGS method was capable of detecting the mutation, correctly defining the breakpoints of the deletion, and provided an accurate estimation of the proportion of mitochondria affected (see online supplementary figure S5). Additionally, the chromosomal status of the cell tested (euploid) was simultaneously confirmed using the same method discussed above. Other research supports the suggestion that mtDNA can be accurately sequenced in small numbers of cells derived from human preimplantation embryos.11
Clinical application of NGS to screen human embryos for aneuploidy, resulting in pregnancies and births
After confirmation of the accuracy of aneuploidy detection in the blinded study outlined above, the NGS method was used to help guide the selection of embryos produced by two infertile couples (women aged 35 and 37 years). A total of five blastocyst stage embryos suitable for biopsy were produced in one IVF cycle and two in the other. A trophectoderm sample (composed of ∼5 cells) was taken from each embryo and tested for aneuploidy using the NGS method. Two embryos were found to have abnormalities, predicted to lead to failure of implantation or early pregnancy loss and were excluded (table 1, samples 57 and 58). The remaining embryos were shown to be euploid. One chromosomally normal embryo, containing typical quantities of mtDNA, was transferred in each instance and resulted in the birth of a healthy baby in the summer of 2013 in both cases. The sex and chromosomal status of the two children were confirmed to be identical to that predicted by NGS.
Discussion
This study describes the design, optimisation, validation and clinical application of an aneuploidy detection method applicable to single cells. The method, based on NGS, was shown to be rapid and highly accurate. Compatibility with utilisation in a clinical setting was demonstrated by the testing of embryos produced using IVF technology, assisting in the identification and transfer to the uterus of chromosomally normal embryos. This resulted in viable pregnancies and the birth of healthy children. Although alternative methods for the detection of aneuploidy in human preimplantation embryos already exist, NGS offers significant advantages in terms of the breadth and scope of the information provided and the cost of testing.
The number of patients requesting preimplantation genetic testing of their embryos is growing significantly. In our own laboratory, we have seen referrals for preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS) rise by over 50% in the last 2 years and many other clinics have witnessed similar increases. The upsurge in the number of cases has intensified the need to develop methods capable of delivering accurate diagnosis of embryos at reduced cost, thereby reducing the financial burden on patients and healthcare systems. The results of the current research confirm that NGS provides highly accurate aneuploidy detection at substantially lower cost than existing methods used for PGS and that testing can be performed within a fresh IVF cycle, avoiding the complication of cryopreservation. A small number of previous studies had suggested that the combination of WGA and NGS might be capable of producing useful genomic data from single cells.12––15 However, previous attempts to harness massively-parallel sequencing methods for the detection of aneuploidy in single cells required several days to complete, meaning that preimplantation genetic testing could only be carried out if embryos were cryopreserved after biopsy. The addition of embryo cryopreservation represents a deviation from the most widely used strategies for embryo analysis, which increases the cost of the procedure and carries a risk of harm to the embryo (most fertility clinics report that 5%–10% of embryos do not survive freezing and thawing).
The need for rapid methods for the genetic testing of preimplantation embryos stems from the fact that the window of time during which implantation can occur is narrow. If embryo biopsy is undertaken at the blastocyst stage (5 days after fertilisation of the oocyte)—increasingly the preferred strategy for PGD and PGS—less than 24 h are available for genetic analysis. In the current study, a protocol requiring under 15 h was used successfully in clinical cycles, a turnaround time similar to speeds achieved using the most rapid microarray-CGH methods currently available and considerably quicker than alternative PGD and PGS methods based on the use of SNP microarrays (see online supplementary table S2). Subsequent work, carried out after the initial clinical cases, demonstrated that reliable aneuploidy detection in single cells biopsied from human embryos is possible in even shorter timeframes (less than 8 h) when using NGS.
As well as the requirement for rapid results, methods used for the analysis of preimplantation embryos must also permit simultaneous testing of multiple samples. Individual patients undergoing IVF/PGD treatment typically produce several embryos, each of which needs to be examined. The fact that each patient is associated with multiple tests is problematic for some PGD and PGS methods: technical bottlenecks mean that equipment used for analysis must be duplicated in order to accommodate all of the samples. NGS has the advantage in that it allows large numbers of samples to be processed concurrently. Up to 32 samples were processed simultaneously during this study and the data obtained suggest that two or three times that number could potentially be multiplexed together without any significant loss of accuracy. Multiplex analysis also has implications in terms of the price of the test. Although, NGS is becoming less expensive every year, at present each run is still associated with an appreciable cost. However, consumable expenses can be shared across several samples tested during the same run using molecular ‘barcoding’ (see online supplementary figure S2). In the current study, this approach allowed NGS to reduce the per-embryo cost by more than a third compared with the most widely used microarray-based approaches (based on 32-plex analysis and manufacturer's list prices). Such a reduction has substantial implications for embryo chromosome screening, strengthening the health-economics argument for clinical utilisation and potentially making it accessible to much greater numbers of infertile couples.
The data presented here provide reassurance concerning the accuracy of aneuploidy detection in cells from preimplantation embryos using NGS. Near complete concordance was achieved in comparison with results obtained using a highly validated aCGH method (see online supplementary tables S5 and S6).4 ,16 A total of 54 samples were tested during the initial validation phase of this study and the diagnosis (normal or abnormal) using the two methods was identical in all cases. Agreement was seen for 99.7% of the 1296 chromosomes evaluated. The only two embryos with any discrepant findings (numbers 34 and 37 in table 1) were each highly abnormal, containing multiple aneuploidies (>3 abnormal chromosomes each). Embryos affected by several chromosome abnormalities frequently display chromosomal mosaicism and consequently a degree of cytogenetic divergence between the different biopsy specimens is expected. In these two cases, most of the aneuploidies were present in both biopsies taken from the same embryo and may have been a consequence of meiotic errors. However, additional aneuploidies were present in one of the two samples. The discrepant findings could be explained by mitotic errors occurring after fertilisation, but this cannot be proven conclusively on this occasion as no further embryo material was available for analysis. The spectrum of abnormalities detected in the embryos analysed during this study are in agreement with the findings of numerous previous investigations, using a variety of cytogenetic techniques, and confirm that a wide range of abnormalities, including types never seen in established pregnancies or miscarriages and presumably lethal during early development, are common prior to implantation.
Diagnosis of mutations responsible for single gene disorders in embryos produced using IVF technology (ie, PGD) is increasingly requested by couples at high-risk of transmitting an inherited disorder to their children.17 In most cases, this involves biopsy of single cells at the cleavage stage, genetic testing and transfer of unaffected embryos to the mother's uterus. Any pregnancies that result following this process should be free of the familial disorder and therefore the issue of selected pregnancy termination is avoided. However, on some occasions the embryos transferred miscarry due to a spontaneously arising chromosome abnormality. This is a particular issue for couples requesting PGD who carry a single gene mutation and are at increased risk of aneuploid conception due to female age. Although the research reported in this paper was principally focused on the use of NGS for preimplantation aneuploidy detection, a pilot experiment was undertaken to assess the feasibility of testing for alterations of DNA sequence in parallel with chromosome screening. Historically, the combination of DNA sequence analysis and chromosome copy number testing in single cells has been challenging. Of the hundreds of protocols for the PGD of gene mutations published in the literature, only a handful are compatible with comprehensive chromosome analysis.18––21 The data presented here are preliminary, but indicate that NGS has the potential to deliver accurate mutation detection as well as aneuploidy screening, overcoming a limitation of traditional PGD tests. It is important to note, however, that rates of allele dropout (failure to amplify one of the two alleles in a heterozygous cell) are appreciable at the single cell level and consequently a significant error rate would be expected for diagnostics based on analysis of the mutation site alone. For this reason, analysis of several flanking polymorphisms remains advisable, supplementing direct mutation detection with redundant diagnoses based on linkage analysis. Strategies using multiple linked markers are commonly used for PGD of single gene disorders.22
While there is accumulating evidence to suggest that aneuploidy is the single most important factor affecting the ability of an embryo to implant and form a viable pregnancy,6––10 ,23 ,24 it remains the case that even the transfer of a chromosomally normal, morphologically perfect embryo to the uterus cannot guarantee a pregnancy. Clearly there are other, less well-defined aspects of embryo biology that are also critical for successful development. A prime candidate in this regard is mitochondrial copy number, which previous studies have linked to various aspects of oocyte and embryo competence.25 ,26 Recent research in our laboratory has demonstrated that an unusually high level of mtDNA in human preimplantation embryos is associated with failure of implantation in the uterus.27 Unlike all other methods of embryo aneuploidy screening currently in use, NGS is capable of simultaneously providing data on mtDNA copy number as well as chromosome imbalance. While further work is needed to replicate our observations, the evidence thus far suggests that quantification of mtDNA may provide an extra dimension to embryo assessment, enhancing our ability to recognise viable embryos and thereby improving the likelihood of successful IVF treatment.27
In the current study, the embryos from clinical cycles that produced babies had typical mtDNA levels, but too few embryos were transferred to allow any conclusions to be drawn concerning the relevance of mtDNA in terms of IVF success. However, an observation of biological and clinical importance was that embryos with high levels of mtDNA had an elevated risk of aneuploidy (p<0.05) (figure 2). The link between chromosomal abnormality and mtDNA content, revealed by NGS, was subsequently confirmed in our laboratory using qPCR and is concordant with a previous study that employed real-time PCR methods.28 It is likely that the detection of greater quantities of mtDNA is indicative of an increased number of mitochondria present in the cells tested, although this remains to be conclusively proven. Raised quantities of mitochondria could lead to abnormally elevated metabolism, a factor that previous studies have speculated might be associated with reduced embryo viability, the so-called ‘Quiet Embryo’ hypothesis.29 Alternatively, expansion of the mitochondrial population could represent a compensatory mechanism, symptomatic of the presence of defective organelles, perhaps a consequence of mtDNA mutation.
The ability of the NGS method described to detect and quantify mtDNA mutation was also demonstrated during this study. mtDNA disorders are responsible for a number of devastating conditions for which there is no cure and few treatment options. In most cases, affected individuals are heteroplasmic, having a mixture of normal and mutant mitochondria in their cells. The severity of the symptoms is determined by the proportion of organelles that are defective. As many as one person in 400 is affected by an mtDNA disorder30 and the phenomenon of the mitochondrial bottleneck means that the phenotype can swing from mild to severe in a single generation. One possibility for the avoidance of mtDNA disorders is to perform PGD, testing cells biopsied from preimplantation embryos and transferring to the uterus only those with entirely normal mitochondria or with low (subclinical) mutation loads.31––37 However, accurate quantification of mtDNA mutations has been technically difficult at the single cell level, requiring extensive protocol design and optimisation for each mutation tested. It seems likely that NGS will provide a single robust method for the detection and quantification of any mtDNA mutation, streamlining the PGD process.
Conclusions
Worldwide, approximately a million assisted reproductive treatment cycles end in failure each year, emphasising the urgent need for improvements to existing techniques. There is mounting evidence from randomised clinical trials that many infertile couples can achieve increased IVF success rates if the embryos chosen for transfer to the uterus have been shown to be euploid, yet only a small fraction of IVF cycles currently include chromosome screening. The wider application of genetic analysis is restrained, in part, by the high costs associated with testing. The technique detailed in this paper has the capacity to cut the cost of analysis significantly and should therefore increase patient access to embryo aneuploidy screening. Unlike previous studies applying NGS to single cells, this method is compatible with the most widely used strategies for embryo testing, which require diagnosis to be completed within ∼24 h of embryo biopsy. The clinical applicability of the test was demonstrated in IVF cycles, resulting in the birth of healthy children. These represent the first births achieved following NGS-based screening of human embryos in the Western hemisphere. The children produced are healthy and developing normally, at 1 year of age.
In theory, NGS methods such as those described here could be extended to allow whole genome sequencing of embryos. While our data and that of other research groups suggest that this is technically feasible, sequencing of the entire genome would greatly increases costs and the time required for analysis, eliminating two of the chief benefits of the strategy described in this paper. Furthermore, knowledge of the genome sequence of a human embryo prior to transfer to the uterus has the potential to reveal unexpected genetic findings that may be difficult to interpret (ie, incidental findings of unknown clinical significance), not to mention the ethical concerns that may be raised by the acquisition of such detailed genetic information and its potential use in embryo selection. The test outlined here deliberately avoided sequencing the whole genome (less than 0.1% was sequenced from each embryo), focusing on low cost, rapid analysis and detection of serious genetic abnormalities that have a well-defined impact on health or embryo viability.
Methods
Samples analysed during preliminary optimisation and validation
Samples were derived from cytogenetically characterised cell lines (Coriell Cell Repositories) or from human embryos previously shown to be aneuploid during routine preimplantation genetic screening (Reprogenetics). Initial analyses began in July 2011, with clinical cases initiated in April 2012. Clinical IVF cycles using NGS took place at Main Line Fertility and New York University Fertility Center. Genetic analyses were carried out at Reprogenetics UK and the University of Oxford.
Single cell lysis
An amount of 2.5 μL of the alkaline lysis buffer (0.75 μL PCR-grade water (Promega, Madison, USA); 1.25 μL dithiothreitol (DTT) (0.1 M) (Sigma, Gillingham, UK); 0.5 μL NaOH (1.0 M) (Sigma)) was added to a 0.2 mL microcentrifuge tube containing a single cell or trophectoderm biopsy (5–10 cells) suspended in 0.5–2.0 μL of phosphate buffered saline. The samples were then incubated for 10 min at 65°C in a thermal cycler.
Whole genome amplification
After lysis, whole genome amplification reagents were then added to each tube (44 μL per sample): 12.5 μL of PCR-grade water (Promega); 2.5 μL of tricine (0.4 M) (Sigma); 29 μL of Repli-g Midi Reaction buffer (Qiagen, Crawley, UK); and 1 μL of Repli-g Midi DNA polymerase (Qiagen). Samples were incubated at 30°C for 120 min and 65°C for a further 5 min.
Optimised NGS protocol
WGA product was quantified using a Qubit (HS dsDNA) and 100 ng fragmented using NEBNext DNA fragmentation enzyme for 17.5 min for Ion Torrent 200 bp chemistry.
Electrophoresis was performed and the sample fraction at 300 bp was excised using E-Gel SizeSelect Gels (Life Technologies).
A minimum of five cycles of adaptor mediated amplification was required to generate quantifiable sequence-ready libraries. The samples were evaluated using a 2100 Bioanalyser High Sensitivity Chip (Agilent Technologies). All samples were diluted to a 24 pmol concentration and a 5 µL aliquot of the sample was used as a template for clonal amplification using the Ion OneTouch system (Life Technologies). Sample enrichment was performed on the Ion OneTouch ES module and Ion 316 chips were used for sequencing. The chip was prepared for sequencing by the use of a modified protocol whereby 30 µL of sample was added between five rounds of 2 min centrifugation steps, resulting in high levels of data generated with each sequencing run.
Data were analysed using the coverageAnalysis (V.2.2.3) plugin in the Torrent Suite V.2.2 (Life Technologies), providing the percentage of DNA sequence reads mapped to each chromosome. The percentage of the reads derived from a given chromosome for an embryo (test) sample was divided by the reference value for the same chromosome as described in the Results section (see also online supplementary table S1). Chromosomal gains were associated with ratios >1.2 and losses with ratios <0.7. For each sample, the number of mtDNA reads was divided by the number of reads attributable to the nuclear genome. This provided an indication of the amount of mtDNA per cell.
Up to 32 samples were multiplexed together and sequenced on the same chip. In order to achieve this, prior to sequencing, amplified DNAs from each sample were fragmented and different adapters of unique DNA sequence (ie, barcodes) from the Ion Xpress Barcode Adapters 1–16 and 17–32 kits were ligated, allowing for postsequencing demultiplexing. The samples were diluted to a concentration of 24 pmol and pooled prior to clonal amplification on the Ion OneTouch system. Semiconductor sequencing of 100 bp was performed on the Ion PGM Sequencer using 200 bp chemistry on a single Ion 316 chip. The fragments sequenced were assigned to specific samples based on their unique barcode.
Procedures for blinded analysis of aneuploidy
A second biopsy was taken from each embryo and coded by a technician who had no other involvement in the study. Analysis of the coded samples was undertaken using a well-established microarray-CGH approach, carried out by scientists in a different laboratory from those performing NGS. The results of both tests (ie, aneuploidy diagnosis using NGS and aCGH) were communicated to an independent adjudicator, samples decoded and diagnostic results compared.
Confirmation of NGS-based mtDNA quantification using real-time PCR
Whole genome amplification products from cells biopsied from blastocyst stage embryos were analysed using real-time PCR. A custom-designed TaqMan Assay (Life Technologies, UK) was used to target and amplify the mitochondrial 16 s ribosomal RNA sequence (probe sequence: AATTTAACTGTTAGTCCAAAGAG). Normalisation of input DNA took place with the reference to a second TaqMan Assay amplifying the multicopy Alu sequence (YB8-ALU-S68) (probe sequence: AGCTACTCGGGAGGCTGAAGGCAGGA). This normalisation was employed to avoid issues such as variation in whole genome amplification efficiency and differences in the number of cells contained in the biopsy specimen. Additionally, a reference DNA sample consisting of amplified 46,XY DNA was included each time real-time PCR was undertaken, allowing variation between experiments to be controlled for. A negative control (nuclease free H2O and PCR master-mix) was also included in all experiments. Alu and mtDNA sequences were amplified in triplicate from each PicoPlex product. Reactions contained 1 μL of whole genome amplified embryonic DNA, 8 μL of nuclease-free H2O, 10 μL of TaqMan Universal Master-mix II (2X)/no UNG (Life Technologies, UK) and 1 μL of the 20× TaqMan mtDNA or Alu assay (Life Technologies, UK), for a total volume of 20 μL. The thermal cycler used was a StepOne Real-Time PCR System (Life Technologies, UK), and the following conditions were employed: incubation at 50°C for 2 min, incubation at 95°C for 10 min and then 30 cycles of 95°C for 15 s and 60°C for 1 min.
PCR amplification of the CFTR gene ▵F508 mutation site
After cell lysis and WGA, a 0.5 μL aliquot was removed from the MDA product and added to 14.5 μL of PCR mixture. The PCR mixture was composed of the following: 1.45 μL 10× Enzyme Buffer (5 Prime, Hamberg, Germany); 0.15 μL of primer mixture (Forward GTTTTCCTGGATTATGCCTGGCAC; Reverse GTTGGCATGCTTTGATGACGCTTC; each at 10 μM); 0.2 mM dNTPs (Sigma); 0.45 units of Hot Master Taq enzyme (5 Prime); and PCR-grade water (Promega). The PCR programme was: 96°C for 1 min; 30 cycles of 94°C for 15 s, 59°C for 15 s, 65°C for 45 s; 65°C for 2 min. This yielded a DNA fragment encompassing the ▵F508 mutation site (95 bp in length if normal, 92 bp if ▵F508 was present). Sequencing libraries were generated from the original WGA product and the resulting PCR product. For clonal amplification and based on template dilution factors calculated according to manufacturer protocol, 1.3 µL of the library from the PCR product (diluted 1:500) was added to 3.7 µL of a 1:16 dilution of the library from the original WGA. The WGA-PCR library mixture was then analysed using NGS as described above.
Microarray-CGH
Microarray-CGH analysis was undertaken according to our previously validated protocol using 24Sure Cytochip (Illumina, Cambridge, UK) (see online supplementary tables S5 and S6). Lysis and whole genome amplification of single cells biopsied from embryos were achieved using the SurePlex kit (Illumina). The entirety of this procedure took place according to the manufacturer's instructions. The fluorescence labelling system (Illumina) was used for the labelling of the amplified ethylenediaminetetraacetic acid-Tris (TE) samples and also for labelling a commercially available reference DNA (Illumina). Test TE samples were labelled with Cy3 while the reference 46,XY DNA was labelled with Cy5. Test and reference DNAs’ co-precipitation, their denaturation, array hybridisation and the posthybridisation washes all took place according to protocols provided by the manufacturer. The hybridisation time was 16 h.
A laser scanner (InnoScan 710, Innopsys, Carbonne, France) was used to analyse the microarrays after washing and drying. The resulting images were stored in TIFF format file and examined by the BlueFuse Multi analysis software (Illumina). Chromosome profiles were examined for gain or loss with the use of a 3× SD assessment.
Clinical application of NGS for preimplantation aneuploidy detection
Two infertile couples (women aged 35 and 37) gave consent for NGS analysis of their embryos following counselling. The protocols used for ovarian stimulation, IVF and embryo culture did not differ from methods considered to be standard. At the blastocyst stage, 5 days after oocyte fertilisation, the zona pellucida encapsulating the embryo was breached with a laser and ∼5 trophectoderm cells were carefully removed using a micromanipulator. The cells were washed in three 5 μL droplets of PBS+0.1% polyvinyl alcohol and then transferred to a 0.2 mL microcentrifuge tube in a total volume of 1.5 μL. Cell lysis, MDA and NGS were carried out as described above. One euploid embryo was transferred in each case, while additional euploid embryos remained cryopreserved (vitrified).
Preliminary evaluation of an ultra-rapid method of NGS-based aneuploidy screening
Subsequent to the validation and clinical application of NGS for the purposes of aneuploidy detection, an even more rapid evolution of the original protocol was evaluated. This involved analysis of 17 samples (∼5 cells each) derived from human blastocysts. In some cases, embryos were biopsied twice, allowing independent testing of cells derived from the inner cell mass and trophectoderm. For all samples, cell lysis and whole genome amplification (MDA) were carried out as described above. This was followed by creation of barcoded libraries from 100 ng of MDA samples using the Ion Xpress Plus protocol described previously with six cycles of amplification. The amplified libraries were run on an EGel SizeSelect 2% agarose gel (Invitrogen) and the fraction corresponding to a 350 bp insert was purified with 0.5× of AMPure beads (Agencourt). Concentrations were estimated by qPCR using the Ion Library Quantitation Kit (Life Technologies) and 1.7 µL of a 50 pM dilution was used as an input for Ion Isothermal Amplification Chemistry (Life Technologies). This generated templated spheres in 30 min. Spheres were then enriched, loaded on an Ion 316 chip (10 samples multiplexed at a time) and sequenced on the Ion PGM for 260 flows using the Ion PGM 200 V.2 Sequencing kit (Life Technologies). Analysis of the DNA sequence reads produced was carried out as described above.
Acknowledgments
This work was supported by the Oxford NIHR Biomedical Research Centre Programme.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All authors had essential roles in this study and each participated in the writing and/or review of the manuscript. DW—study concept, laboratory work, manuscript preparation; KK—laboratory work; JG and MG—patient counselling and treatment (IVF); JCT, EF, SM—genetics support, data analysis.
-
Competing interests None.
-
Ethics approval NRES Committee South Central.
-
Provenance and peer review Not commissioned; externally peer reviewed.