Article Text

Abstract
Background The most common spinocerebellar ataxias (SCA)—SCA1, SCA2, SCA3, and SCA6—are caused by (CAG)n repeat expansion. While the number of repeats of the coding (CAG)n expansions is correlated with the age at onset, there are no appropriate models that include both affected and preclinical carriers allowing for the prediction of age at onset.
Methods We combined data from two major European cohorts of SCA1, SCA2, SCA3, and SCA6 mutation carriers: 1187 affected individuals from the EUROSCA registry and 123 preclinical individuals from the RISCA cohort. For each SCA genotype, a regression model was fitted using a log-normal distribution for age at onset with the repeat length of the alleles as covariates. From these models, we calculated expected age at onset from birth and conditionally that this age is greater than the current age.
Results For SCA2 and SCA3 genotypes, the expanded allele was a significant predictor of age at onset (−0.105±0.005 and −0.056±0.003) while for SCA1 and SCA6 genotypes both the size of the expanded and normal alleles were significant (expanded: −0.049±0.002 and −0.090±0.009, respectively; normal: +0.013±0.005 and −0.029±0.010, respectively). According to the model, we indicated the median values (90% critical region) and the expectancy (SD) of the predicted age at onset for each SCA genotype according to the CAG repeat size and current age.
Conclusions These estimations can be valuable in clinical and research. However, results need to be confirmed in other independent cohorts and in future longitudinal studies.
ClinicalTrials.gov, number NCT01037777 and NCT00136630 for the French patients.
- Movement disorders (other than Parkinsons)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Autosomal dominant cerebellar ataxias, also known as spinocerebellar ataxias (SCA), are neurodegenerative diseases that are clinically and genetically heterogeneous. Major advances have been made in the understanding of their causes since the 1990s and mutations in more than 20 genes have been identified thus far to be responsible for different forms of the disease. These mutations are comprised of conventional mutations, non-coding nucleotide expansions, and coding (CAG)n expansions.1 SCA1, SCA2, Machado-Joseph or SCA3, SCA6, SCA7, SCA12, SCA17, and dentatorubral-pallidoluysian atrophy (DRPLA) are caused by (CAG)n repeat expansions in the ATXN1, ATXN2, ATXN3, CACNA1A, ATXN7, PPP2R2B, TBP, and ATN1 genes, respectively, and all lead to the expansion of a polyglutamine tract in the corresponding proteins. Repeat-associated non-ATG translation (RAN) of polyglutamine tracts has also been observed in SCA8 and may contribute to the disease process.2 All so-called polyglutamine ataxias share many common features, including a negative relationship between age at onset and the number of repeats in the expansion, and a more severe disease with larger expansions. The mean age at onset of symptoms for SCA1, SCA2, SCA3, and SCA7 carriers is generally in the third or fourth decade of life, but an average of 20 years later for SCA6 carriers.3 The threshold of CAG expansions, or the number of expansions that determines disease carrier status, varies between the different forms of SCA as do the boundaries between what is considered an expanded and normal size (overlapping in SCA1). In most forms this threshold can be found around 40 repeats, except for in SCA6 where it is closer to 20.1
Gait ataxia is the first symptom identified in the majority of cases of these diseases. Globas et al4 have shown that only 12% of SCA1, 13% of SCA2, 15% of SCA3, and 24% of SCA6 patients have other symptoms before the onset of gait ataxia. Nevertheless, the onset and the phenotype may differ considerably between two individuals with the same genotype.5 Previous studies investigating the relationship between CAG repeat length and age at onset are of limited use in predicting the mean age at onset, as they have relied on simple linear correlations in patients and did not build predictive models that take into account information from clinically unaffected mutation carriers, thus creating a bias favouring pathology. In another polyglutamine disease, Huntington's disease, similar modelling has been performed using statistical models that elucidated the relationship between CAG length and age at onset.6–8 In SCA, a similar approach was used in the Cuban SCA2 population,9 although this approach has not been repeated in other forms of SCA.
It is crucial that studies dealing with prediction of disease onset include both affected individuals and preclinical individuals, which has not been the case in previous models. Ignoring individuals who are free of disease symptoms, are the same age, and have the same number of CAG repeats as affected individuals creates an artificial tendency towards earlier disease onset. For the purposes of this study, we pooled genetic and age at onset data of a large group of SCA1, SCA2, SCA3, and SCA6 patients from the European EUROSCA registry with data of clinically unaffected carriers of SCA1, SCA2, SCA3, and SCA6 mutations from the RISCA study. The EUROSCA registry was established in 2004 to collect core data of European SCA patients. RISCA is a prospective, multicentric, multinational, observational cohort of clinically unaffected at-risk individuals for SCA1, SCA2, SCA3, and SCA6 (ie, first degree relatives of patients with one of these diseases).10
Patients and methods
Patients
Two groups of individuals were included: affected patients (EUROSCA registry) and preclinical mutation carriers (RISCA cohort). The EUROSCA registry includes individuals with any form of spinocerebellar ataxia (SCA) from 17 European centres. For the current study, we selected 1187 patients with a positive molecular genetic test for SCA1, SCA2, SCA3 or SCA6, genotyped at a central laboratory, and with information available on age at onset of the disease (317 SCA1, 308 SCA2, 399 SCA3, and 163 SCA6) and, when possible, a SARA (Scale for the Assessment and Rating of Ataxia, with a maximal score of 40 indicating a very severe cerebellar ataxia) score ≥3.10 Patients were included in the database with age at onset as indicated by self-report during their examination by the neurologist, and as indicated in their medical records. Disease onset was defined by the onset of gait difficulties, as this is the most frequent first symptom. Data were obtained from patients by personal interview. Information obtained by interview was then compared to that from medical records, if available.
The RISCA cohort included individuals at-risk for SCA from 14 European centres.11 These included adult individuals, children or siblings of an individual with SCA1, SCA2, SCA3 or SCA6. Absence of ataxia was defined as having a score on the SARA scale <3. All individuals were genotyped in the same central laboratory as the EUROSCA registry, and of the 264 individuals included with DNA available, 123 (47%) were carriers of a disease-causing expansion (50 SCA1, 31 SCA2, 26 SCA3, and 16 SCA6). For these preclinical mutation carriers, the age at examination was recorded.
All participants signed informed consent documents approved by institutional review boards and the local ethics committee.
Genotypes
Blood samples to obtain DNA for genetic testing were taken from all study participants including those who had already undergone preclinical genetic testing. All genetic tests were performed at the Institute of Medical Genetics and Applied Genomics (Tübingen, Germany) using established and standardised methods.
For the RISCA cohort, the genetic tests were done anonymously under an arrangement that guaranteed that results were not disclosed to study participants, clinical investigators or anyone else except the statistician's team (STdM and ID-G). However, all study participants were offered genetic counselling with an open preclinical testing procedure according to established clinical standards.
For both cohorts, we defined the pathological thresholds as a CAG repeat expansion of more than 39 repeats in SCA1, more than 31 repeats in SCA2, more than 47 repeats in SCA3, and more than 20 repeats in SCA6.
Prediction of age at onset
Statistical model
The prediction of age at onset was achieved using a statistical model to relate the age at onset of an individual with his genotype. As our final sample included some individuals who had not yet reached an age to be affected by the disease but will inevitably develop symptoms, the methodological framework we used was one of survival analysis. In order to make predictions about age at onset, we used a parametric survival model, namely a log-normal censored model. The age at onset was predicted from the moment of birth for a patient with known genotype using the following formulae: 1where: T is the age at onset from birth, a random variable for a patient with known genotype,
1where: T is the age at onset from birth, a random variable for a patient with known genotype,  is the expectation of Log(T),
is the expectation of Log(T),  is the SD of Log(T), and E is a random variable with a standard Gaussian probability density function.
is the SD of Log(T), and E is a random variable with a standard Gaussian probability density function.
The mean log age at onset  , for a given genotype, is derived from a regression model as follows using the numbers of repeats of the two alleles ne and nne: expanded and not expanded respectively:
, for a given genotype, is derived from a regression model as follows using the numbers of repeats of the two alleles ne and nne: expanded and not expanded respectively: 2where
2where  ,
,  and
and  are the regression parameters that are being estimated.
are the regression parameters that are being estimated.
The random variable T (age at onset) has a log-normal probability density function (pdf) with parameters  and
and  . Thus its pdf is:
. Thus its pdf is: 3
3
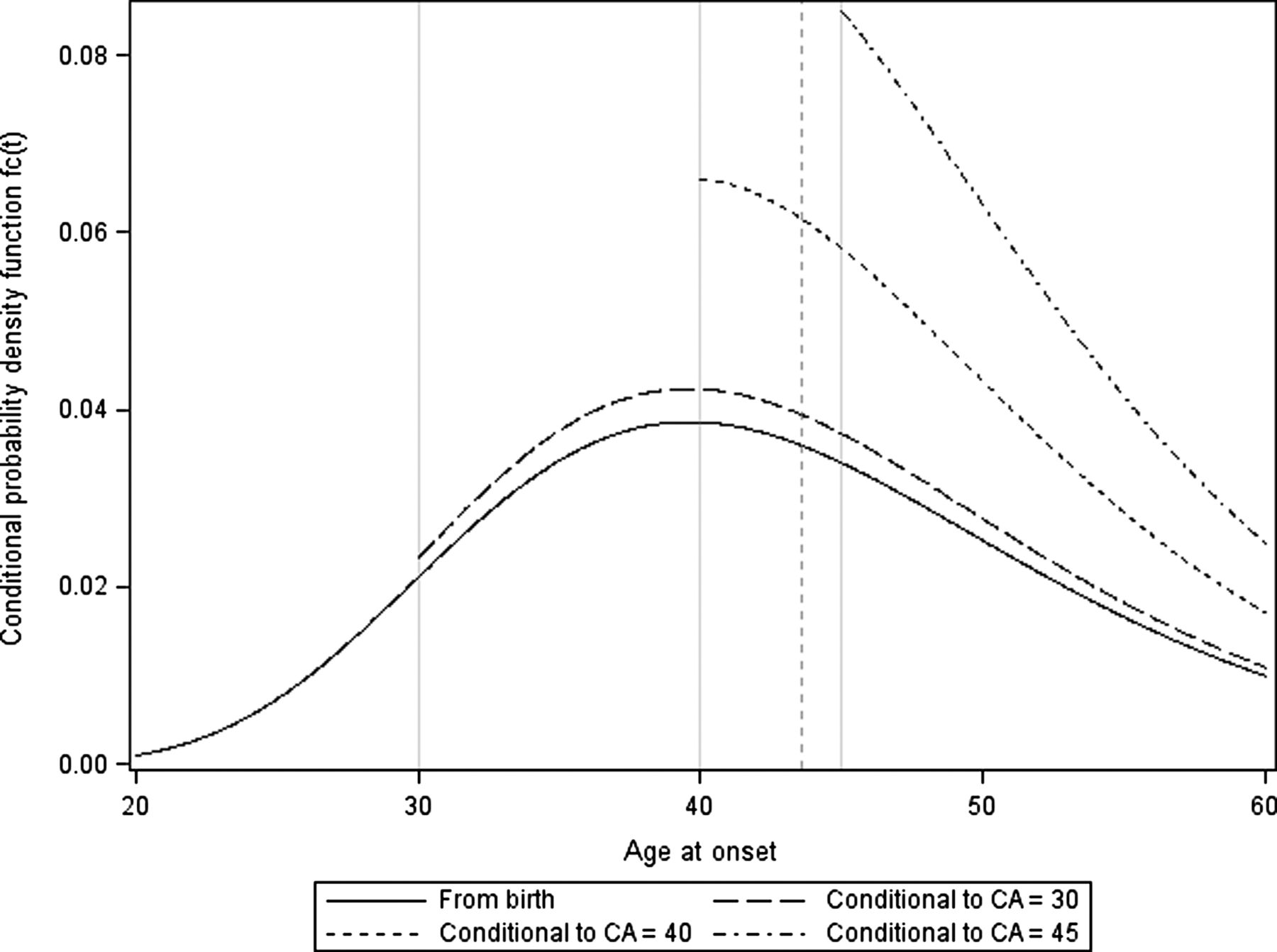
As an example f(t) is plotted for SCA2 with a repeat number of the expanded allele of 37 (figure 1).

Conditional probability density function of age at onset for different current ages (CA) (t). The figures are drawn for SCA2 data with a repeat number of the expanded allele of 37 for an individual at birth (solid), or at a current age of 30 (medium dash), 40 (short dash) or 45 (short dash dot). Based on a log linear parametric model, the estimation of the mean age at onset from birth was 43 years (γG: 3.7428, : 0.2516) (vertical dashed line). As the distribution is log-normal the mode is neither the mean nor the median. SCA, spinocerebellar ataxias.
: 0.2516) (vertical dashed line). As the distribution is log-normal the mode is neither the mean nor the median. SCA, spinocerebellar ataxias.
When T is censored, we need to express S(t)=P(T>t). We have:
S(t)=1—F(t), where 4
4
And finally: 5
5
Estimation of the parameters
The estimation of the parameters was performed using the affected individuals from the EUROSCA registry whose age at onset is known, and the unaffected individuals carrying an expanded allele from the RISCA study. For the latter patients, the age at last examination was known, and we considered that this age was a censored value of the age at onset of the disease. The parameters were estimated by the maximum likelihood method. Backward selection was used to retain the significant parameter  or
or  . The parameter estimation of the model was performed using the SAS V9.3 statistical software.
. The parameter estimation of the model was performed using the SAS V9.3 statistical software.
Computation of the predictive statistics: expectation, SD, and percentiles
In order to take into account that we used parameter estimates, we added to the variance  the variance of
the variance of  , estimated from the estimated parameters
, estimated from the estimated parameters  ,
,  and
and  , with
, with 6
6
And finally: 7where, var(
7where, var( ), var(
), var( ) and var(
) and var( ) are the variances of the estimated parameters
) are the variances of the estimated parameters  ,
,  and
and  respectively (table 1), cov(
respectively (table 1), cov( ,
,  ), cov(
), cov( ,
,  ) and cov(
) and cov( ,
,  ) are the covariances between the estimated parameters (see online supplementary table S1).
) are the covariances between the estimated parameters (see online supplementary table S1).
Parameter estimates obtained from the parametric survival model
We thus computed the predictive statistics from the estimated pdf of the age at onset, from birth or conditionally that this age is greater than the current age. The estimated pdf of the age at onset is given by the formulae: 8where
8where  is given by formula (2) after replacing the parameters by their estimates and
is given by formula (2) after replacing the parameters by their estimates and  is given by formula (7).
is given by formula (7).
From these formulae, one can derive the values of the predictive statistics (expectation E(t), variance var(T), and percentiles tα) of the age at onset distribution. These predictive statistics are calculated first from the moment of birth, without regard to the actual disease progression of the individual.
We have: 9and
9and 10The
10The  th percentile
th percentile  is thus obtained from the inverse of F such as:
is thus obtained from the inverse of F such as: 11
11
In order to account for the fact that any given asymptomatic individual has reached his current age c without yet being affected by disease, we thus estimated the age at onset given a current age (c). As shown in figure 1, this leads to a truncation of the log-normal distribution which increases with c. As the individuals are not observed at birth, but at a current age c, we need to estimate E(T|T>c), the expectation of T given that the individual's age is more than c, the corresponding variance Var(T|T>c) and the corresponding percentiles tα.
 12
12
 13
13
with
 14And the ath percentile is given by:
14And the ath percentile is given by:
 15
15
In this paper, we computed the 5th, 50th (median) and 95th percentiles of the T pdf. We called the (5th; 95th) interval the critical region (90% CR).
Model validation
We conducted a validation study in order to assess the goodness-of-fit of the log-normal model. For each type of SCA disease, the model's validation is based upon the comparison of the observed survival function of the whole sample, as obtained by the Kaplan-Meier method, and the sample estimated survival function. The sample estimated survival function was obtained by the crossover method: for each individual of the sample, the parameters of the model were obtained by removing the individual from the sample, and estimating the survival function of the individual based on its genotype. The sample estimated survival function is the mean of these estimated survival functions for each individual.
Sensitivity analysis
One limitation of our study is that the sample we used was obtained by merging two samples, one with affected patients and one with preclinical mutation carriers. As discussed previously in this paper, while it is crucial to include both affected and unaffected carriers, the two samples do not have the same parameters and thus the accuracy of our results may depend of the respective proportions of the two populations. In order to study the sensitivity of the results to these proportions, we conducted a sensitivity analysis with the following method: we modified the proportions of the two sub-samples by multiplying the unaffected sample size by the factors 0.5 (half of the unaffected) and 2 (twice as many unaffected). This was done by giving these weights to each individual within the preclinical mutation carrier sample, and by making all computations with these weighted samples.
Results
Description of the populations
We included 1310 individuals; of these 1187 were EUROSCA affected individuals (SCA1: 317, SCA2: 308, SCA3: 399, SCA6: 163) from 735 families and the remaining 123 were RISCA unaffected individuals (SCA1: 50, SCA2: 31, SCA3: 26, SCA6: 16) from 102 families. Forty-two families included both EUROSCA affected (120 individuals) and RISCA unaffected individuals (51 individuals). Half of the individuals were males, and half were females. SCA6 individuals were older than the individuals from the other genotypes. As expected, within each genotype, the mean age at last examination for the unaffected individuals was lower than the mean age at onset of the affected individuals (SCA1: <0.0001, SCA2: 0.0047, SCA3: 0.0051, SCA6: 0.0287). However, there was overlap as the age of some unaffected individuals was higher than the age at onset of some affected individuals (table 2).
Characteristics of affected and unaffected individuals according to their spinocerebellar ataxias (SCA) genotype
Parametric model
For SCA2 and SCA3 genotypes only the number of repeats of the expanded allele was significantly associated with the age at onset, while for SCA1 and SCA6 genotypes the number of repeats of both alleles were significantly associated (table 1) with age at onset. The recruiting centre, family, and year at onset separated as quartile did not substantially influence the results. For all genotypes, gender was not significantly associated with age at onset, but, as expected, the expanded allele had a negative effect on the age at onset. For SCA1, the log of the age at onset decreased by 0.049±0.002 (SE) (p<0.001) for each additional repeat, for SCA2 by 0.105±0.005 (p < 0.001), for SCA3 by 0.056±0.003 (p<0.001), and for SCA6 by 0.090±0.009 (p<0.001). In addition, in SCA1, the log of the age at onset increased by 0.013±0.005 (p=0.014) with each additional repeat on the shorter non-expanded allele, and in SCA6, the log age at onset decreased by 0.029±0.010 (p=0.0075).
Prediction of age at onset
Based on a log-normal distribution of the age at onset, we obtained the age at onset for each genotype and the range of observed repeat lengths within each genotype. For example, an individual with 37 repeats in the SCA2 gene would have a median age at onset of 42 years old (90% CR: 28–64) (figure 1, see online supplementary table S3). Given that this individual is unaffected at the age of 35 years, he would have a 50% risk of developing the disease before the age of 45 years (90% CR: 36–66) and if he remains unaffected at the age of 45 years, he would have a 50% risk of onset before the age of 52 years (90% CR: 46–71) (figure 2B, see online supplementary table S3). Similar results were obtained for SCA1 (figure 2A, see online supplementary table S2), for SCA3 (figure 2C, see online supplementary table S4), and for SCA6 (figure 2D, see online supplementary table S5). For all SCAs, the accuracy of prediction of the age at onset decreased with the size of the allele expansion: for those with large repeat expansions prediction was more accurate compared to those with mildly expanded alleles. In addition, on average, only 4% of the variance of the age at onset (from 1% for SCA3 to 10% for SCA6) was due to the precision of the statistical model estimation, the remaining being due to population dispersion.

{kind=link}
{kind=link}
Median age at onset according to the genotype and the current age of the presymptomatic individual. (A) SCA1 genotype, (B) SCA2 genotype, (C) SCA3 genotype, (D) SCA6 genotype. For all panels, the x axis is the number of repeats for the expanded alleles, and the y axis the estimated age at onset. For SCA1 and SCA6 genotypes, each sub-panel representing different repeats numbers of the shorter allele are depicted. Curves are plotted from birth and for an individual of 25/30/35/40/45 years old. SCA, spinocerebellar ataxias.
The models were fitted to the observed data (see online supplementary figure S1). Furthermore, the models were robust with respect to the proportion of censored data (see online supplementary figure S2).
Discussion
Using two unique cohorts (the EUROSCA and RISCA cohorts) comprised of individuals recruited at the same European centres, examined by the same clinicians and genotyped in the same centralised laboratory, we were able to estimate the relationship between the number of CAG repeats and the age at onset of gait ataxia in the genes corresponding to the four most frequent polyglutamine ataxia diseases: SCA1, SCA2, SCA3, and SCA6.
Disease onset as defined by the onset of gait difficulties can be variable among patients, and may also be variable depending on the presence of other patients within a given family, as the other members are likely to pay closer attention to early disease symptoms. In contrast to other neurodegenerative diseases such as Huntington's disease, in SCAs there are no psychiatric changes or anosognosia that could interfere with the identification of onset by the patient or their families. Estimations were made according to the genotype of the major gene (number of repeats of the expand allele and additionally, for SCA1 and SCA6, the number of repeats in the short allele) and according to the current age of the carrier of an expansion. There are two events necessary for the disease to develop—the presence of an abnormal CAG repeat, and advanced age. For the mutation carriers with the longest expansions, the oldest age of onset estimations are 1 or 2 years later than the current age. For these carriers, very few individuals will be unaffected in old age, therefore estimations of onset at the oldest ages are more theoretical than real. The ages at onset estimated in this study were similar to those observed and were dependent on CAG repeat length.12 ,13 In addition, as observed, the estimations’ variability decreases with the number of repeats, the smaller the repeat the more accurate the onset estimation. Small but important contributions of the normal polymorphic expansion on the unaffected allele were identified in SCA1 and SCA6, but not in SCA2 and SCA3.3 Similarly to Van de Warrenburg et al, we found a positive effect of the non-expanded CAG repeat for SCA1 and a negative effect for SCA6. This result must be confirmed in an independent cohort, as slightly less than a third of the affected individuals of the current study were also in the Van de Warrenburg et al study.
As has been done previously in Huntington's disease,6–8 we included both affected and unaffected carriers of the mutation. If only affected subjects had been used this could have introduced a bias. Healthy carriers of an abnormally expanded repeat could be different from affected carriers of the same age. In particular, individuals with abnormal expansions in the range just above normal size are expected to start the disease late in life; consequently, they have competing mortality risk and could die of other diseases before the onset of ataxia. This is particularly relevant for SCA6 which has the latest onset of all SCAs. Using only data of affected individuals for estimating the influence of the size of the CAG repeat could lead to a bias providing unduly pessimistic estimates of age at onset.7 To avoid these biases, a survival analysis allowed us to take into account the unaffected but censored individuals. Almaguer-Mederos et al9 published the mean and median age at onset from birth for a range of CAG expansion sizes in SCA2 mutation carriers from a Cuban founder population. Compared to their results, our estimates produced a younger age at onset in SCA2. However, the Cuban estimations were not corrected for the current age of the patients. Even when we applied the same methodology —that is, Kaplan-Meier estimates stratified by repeat length—our estimations of age at onset were lower for each repeat length than those in the Cuban population (data not shown). This may be due to specific properties of the founder population of Cuba or to different recruitment strategies. This was also observed in Huntington's disease in the Venezuelan population as reported by the US–Venezuela Collaborative Research Project and Nancy S Wexler.14 Both the Venezuelan Huntington cohort and the Cuban SCA sample contained affected and preclinical carriers, from large families with a homogeneous genetic background. Their results could be due partly to a specificity of the population, for example, a modifier gene or an environmental effect present in the Cuban population but absent from our study sample population. The samples of our study were recruited in a two-step procedure: first, the affected subjects, and then, their unaffected relatives without systematic screening of the families. Because of this, there may be some carriers within these families with subclinical signs that were not included in either the affected or unaffected cohorts. This could have led to pessimistic estimations of onset age as not all unaffected expansion carriers are necessarily included. Conversely, we have shown that the inclusion of some additional unaffected carriers would have only a small impact on the estimations.
The range of repeat lengths did not cover the entire range that has been previously published. Thus, our results are only valid and usable within this smaller range. An extrapolation outside the range of observed repeats would be misleading. In addition, the present results need to be confirmed either in a replication cohort, or by longitudinal data. These data are not currently available. In addition, the subjects included in the EUROSCA and RISCA cohorts are of primarily European origin. Thus, the extension of the results to other geographical origins must be done cautiously.
Both the SDs of age at onset and the critical regions of the predicted ages—the interval where we have a 90% chance to have the observed age—were quite large. Most of the estimated age variance comes from age dispersion within the population, so it cannot be significantly decreased by a larger sample size. The use of these estimates for clinical purposes, particularly in the context of predictive testing, must be done very carefully, taking into account the variability of the estimates. Keeping these limitations in mind, the estimations can be of help when counselling presymptomatic carriers for the patient that requests it. One risk of this kind of use could be that the knowledge of one's expected age at onset might induce an earlier onset for carriers that are aware of their genetic status. However, data from Huntington's disease do not seem to confirm this kind of effect. In a cohort of presymptomatic Huntington carriers, knowledge of one's genetic status after presymptomatic testing did result in increased auto-observation, but the onset of this disease has always been difficult to define for the carrier and the care taker, as psychiatric symptoms and anosognosia can complicate the determination of disease onset.15 In the case of SCAs this estimation could be more accurate as anosognosia is not present in this disease. In addition, the estimates can be used for epidemiological purposes—for example, to correlate the time to onset to a particular clinical phenotype such as the score on a disease rating scale or to associated phenotypes such as cerebral imaging results. Knowing the expected age at onset in preclinical individuals, Jacobi et al10 were able to infer that for SCA1 and SCA2 mutation carriers the extent of functional and brain structural alterations increased as the interval to the predicted age of ataxia onset decreased.
Acknowledgments
We sincerely thank all the patients and their families for their participation. Caterina Mariotti thanks the AISA-Association (Associazione Italiana Sindromi Atassiche, Sezione Lombardia). We are grateful to Drs Elzbieta Zdzienicka and Rafal Rola (Institute of Psychiatry and Neurology, Warsaw, Poland) for contribution of patients and help in patient's assessment. Many thanks to Sarah Boster for critical reading.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors STdM and TK conceived the study; STdM and J-LG established the prediction of age at onset formulae and calculations. STdM and ID-G performed the analysis. TK and AB contributed to the financing and administration of the registry and cohorts. SF, HJ and GS managed the clinical and genetic data. PB and AS performed the genetic testing. AD, MR, LN, PC, CaM, RR, LS, HJ, SF, TSH, AF, DT, BPvdW, CeM, JSK, PG, AC, LB, MB, SB, SS, JB, JI, KB, MM, RDF, CD, SR, TK and AB enrolled the participants. The manuscript was drafted by STdM and JLG. All authors contributed to the final version of the paper. All authors approved the final version.
-
Competing interests TK receives/has received research support from the Deutsche Forschungsgemeinschaft (DFG), the Bundesministerium für Bildung und Forschung (BMBF) and the European Union (EU). He serves on the editorial board of Parkinsonism and Related Disorders and The Cerebellum. He received a lecture honorarium from Lundbeck. He receives royalties for book publications from Thieme, Urban & Schwarzenberg, Kohlhammer, Elsevier, Wissenschaftliche Verlagsgesellschaft Stuttgart and M. Dekker. MR received a grant as Principal Investigator from the Polish Ministry of Science and Higher Education (Grant No 674N-RISCA/2010–2014); as co-Investigator from the Polish Ministry of Regional Development Operating Programme Innovative Economy (POIG 01.01.02-14-051/09/01—Molecular basis and attempt to genetic classification of patients with clinical symptoms of spastic paraplegia). AS received the following grants: Co-Investigator Grant No 674N-RISCA/2010—2014 from Polish Ministry of Science and Higher Education; Principal Investigator—Molecular basis and attempt to genetic classification of patients with clinical symptoms of spastic paraplegia.—POIG 01.01.02-14-051/09/01- Ministry of Regional Development Operating Programme Innovative Economy. (2010-2013); Co-Investigator: “Molecular analysis in neurodegenerative diseases caused by dynamic mutations” PL0076, 2007–2010, supported by Norwegian Financial Mechanism. Principal Investigator—Analysis of DNA sequence and RNA structure of microsatellite CTA/CTG repeats region in ATXN80S gene and the attempt to explanation of reduced penetrance phenomenon of dynamic mutation causing spinocerebellar ataxia type 8”. N401 097536 ; 0975/B/PO1/2009/36—(2009-2012) Polish Ministry of Science and Higher Education. TSH receives/ has received funding from the European Union (EUROSCA project) and Deutsche Forschungsgemeinschaft (DFG, Klinische Forschergruppe Tiefe Hirnstimulation KFO 247).
-
Funding This study was supported by grants EUROSCA/LSHM-CT-2004-503304 from the European Union, grant from the European Community's Seventh Framework Programme (FP7/2007-2013 n° 2012-305121 NEUROMICS), GeneMove/01 GM 0503 from the German Ministry of Education and Research, within the framework of the ERA-Net for Research Programmes on Rare Diseases, grant 3 PO5B 019 24 from the Polish Ministry of Scientific Research and Information Technology, and grant No 674N—RISCA/2010—2014 from Polish Ministry of Science and Higher Education. The research leading to these results has received funding from the programme ‘Investissements d'avenir’ ANR-10-IAIHU-06. BPvdW is supported by research grants from the Netherlands Brain Foundation, the Royal Dutch Society for Physical Therapy, BBMRI-NL, and the Radboud University Medical Centre. MB is supported by the grant OTKA K 103983. AF was supported by a grant from POR CREME 2007-20013. AB was supported by the grant EUROSCA/LSHM-CT-2004-503304 and by the grant NEUROMICS (7th framework programme) from the European Union. AB, AD and GS received funding from the VERUM Foundation. AD received support from ANR (French Research Agency) and Eranet for the Risca project, PG and AC work at University College London Hospitals/University College London which receives a proportion of its funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme. Paola Giunti receives funding from the EC (HEALTH-F2-2010-242193; FP7 Grant). DeNDRoN, Ataxia UK and NIHR, Department of Health.
-
Patient consent Obtained.
-
Ethics approval Institution review boards and ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction