Article Text

Abstract
Lynch syndrome (LS) is an autosomal dominant disorder caused by a defect in one of the DNA mismatch repair genes: MLH1, MSH2, MSH6 and PMS2. In the last 15 years, an increasing number of patients have been described with biallelic mismatch repair gene mutations causing a syndrome referred to as ‘constitutional mismatch repair-deficiency’ (CMMR-D). The spectrum of cancers observed in this syndrome differs from that found in LS, as about half develop brain tumours, around half develop digestive tract cancers and a third develop haematological malignancies. Brain tumours and haematological malignancies are mainly diagnosed in the first decade of life, and colorectal cancer (CRC) and small bowel cancer in the second and third decades of life. Surveillance for CRC in patients with LS is very effective. Therefore, an important question is whether surveillance for the most common CMMR-D-associated cancers will also be effective. Recently, a new European consortium was established with the aim of improving care for patients with CMMR-D. At a workshop of this group held in Paris in June 2013, one of the issues addressed was the development of surveillance guidelines. In 1968, criteria were proposed by WHO that should be met prior to the implementation of screening programmes. These criteria were used to assess surveillance in CMMR-D. The evaluation showed that surveillance for CRC is the only part of the programme that largely complies with the WHO criteria. The values of all other suggested screening protocols are unknown. In particular, it is questionable whether surveillance for haematological malignancies improves the already favourable outcome for patients with these tumours. Based on the available knowledge and the discussions at the workshop, the European consortium proposed a surveillance protocol. Prospective collection of all results of the surveillance is needed to evaluate the effectiveness of the programme.
- CMMR-D
- Constitutional mismatch repair deficiency
- Surveillance
- Guidelines
- Tumour spectrum
Statistics from Altmetric.com
Introduction
Lynch syndrome (LS) is an autosomal dominant disorder caused by a heterozygous defect in one of the DNA mismatch repair (MMR) genes, that is, MLH1, MSH2 (and EPCAM deletion-mediated MSH2 methylation), MSH6 or PMS2. Carriers of a MMR defect have a high risk of developing colorectal cancer (CRC), endometrial cancer and various other cancers, most of which are diagnosed between the ages of 40 years and 60 years.1 In the last 15 years, an increasing number of patients have been described with biallelic MMR gene mutations in which MMR defects are inherited from both parents. This leads to a syndrome with recessive inheritance, which is referred to as ‘constitutional mismatch repair-deficiency’ (CMMR-D). The spectrum of cancers observed in patients with this syndrome differs from the spectrum found in LS,2 as about half develop brain tumours (BTs), around half develop digestive tract cancers and a third develop haematological malignancies. LS-associated tumours such as endometrial and urinary tract cancers also occur. A large proportion (up to 40%) of patients with CMMR-D, even more than in LS, develop metachronous second malignancies.2 The prognosis of CMMR-D is much worse than that of LS due to the type of malignancies that occur and the high risk of second primary tumours.
BTs and haematological malignancies are mainly diagnosed in the first decade of life, and CRC and small bowel cancer (SBC) in the second and third decades of life. Endometrial cancers and urinary tract cancers are diagnosed in young adult patients with CMMR-D. A variety of non-malignant lesions may also be observed in CMMR-D, such as cafe au lait spots and other signs reminiscent of neurofibromatosis type-I, hypopigmentation, mild immunoglobulin deficiencies and congenital malformations.
Surveillance for CRC in patients with LS is very effective, as regular colonoscopy has been shown to reduce CRC-associated mortality by more than 60%.3 Therefore, an important question is whether surveillance for the most common CMMR-D-associated cancers might also be effective. In view of the diverse nature of the malignancies associated with the syndrome, it is not clear whether early detection is possible and will improve the prognosis.
Recently, Durno et al4 published the outcome of a surveillance programme of two sisters with CMMR-D. Fifteen tumours were detected over a follow-up period of 10 years, including a jejunal carcinoma and a small asymptomatic anaplastic astrocytoma that could be completely resected. Sjursen et al5 reported on a patient who was followed with upper and lower endoscopy, CT and MRI at regular intervals over a period of 26 years. During this time six adenocarcinomas (of the left colon, the duodenum, the distal ileum and the proximal ileum, the proximal jejunum and the endometrium, respectively), as well as several polyps of the large and small bowels and the stomach were removed.
More than four decades ago, a WHO proposal defined the criteria that should be met prior to implementation of large scale population screening.6 These criteria can also be applied in the assessment of surveillance of individuals with a genetic predisposition to cancer. The most important criteria include: (A) cancer should be a common problem in the group targeted for surveillance; (B) the natural course of the cancers should be known; (C) screening tests with high sensitivity and specificity should be available and the tests should be acceptable to the patient; (D) an effective treatment should be available following detection of a tumour; (E) there should be evidence that screening leads to diagnosis of cancer at an early stage and to an improvement in prognosis; and finally, (F) the surveillance protocol should be cost-effective.
Recently, a new European consortium was established with the aim of improving care for patients with CMMR-D. At a workshop of this collaborative group held in the Saint-Antoine Hospital, Paris, (9th of June, 2013), one of the issues addressed was the development of surveillance guidelines. A total of 20 experts in the field, including human and clinical geneticists, pathologists and paediatric oncologists from five countries, participated in the meeting. Experts in the field not present at the meeting were also involved in the discussions on surveillance. In this manuscript, the most important WHO surveillance criteria are addressed, and then applied to assess surveillance in CMMR-D. Based on the outcome of the workshop and the recommendations of European experts, the consortium now proposes a surveillance programme. In view of the lack of studies other than case reports, we did not use a system to grade the category of evidence of reported studies and/or strength of the recommendations.
What is the tumour spectrum in CMMR-D?
A total of 91 families with CMMR-D, including 146 patients, were identified in the world literature (Wimmer et al, in preparation). The most frequent underlying gene defects were PMS2 mutations, which were reported in approximately 60% of cases. The remaining 40% of cases were equally distributed among MSH6 and MLH1/MSH2 biallelic mutation carriers. The various cancers observed are summarised in table 1.
Overview of the most common cancers observed in 146 patients with CMMR-D (Wimmer et al, in preparation)
BTs and digestive tract cancers were the most common cancers, identified in 53% and 40% of patients, respectively. Among the BTs, the most common cancer was high grade glioma, followed by primitive neuroectodermal tumours (PNETs) and medulloblastoma. Glioblastomas and other high grade gliomas were much more frequent in CMMD-R than would be expected based on the relative contribution of these BTs to total paediatric malignancies in the general population. Digestive tract cancers included CRC (40% of all CMMR-D cases) and SBC (12% of all cases). Haematological cancers were observed in 31% and included mainly T cell non-Hodgkin lymphoma (NHL) and acute leukaemia. Other LS-associated cancers (endometrial cancer, urinary tract cancer) were also observed in adults. In addition, a large variety of other tumours were found (table 2). The distribution of ages at diagnoses for BT, haematological malignancies, CRC and SBC reported in the literature are shown in figures 1⇓⇓–4. It should be emphasised that the age distribution is biased by collection of published cases.
Other tumours observed in 146 patients with CMMR-D (Wimmer et al, in preparation)

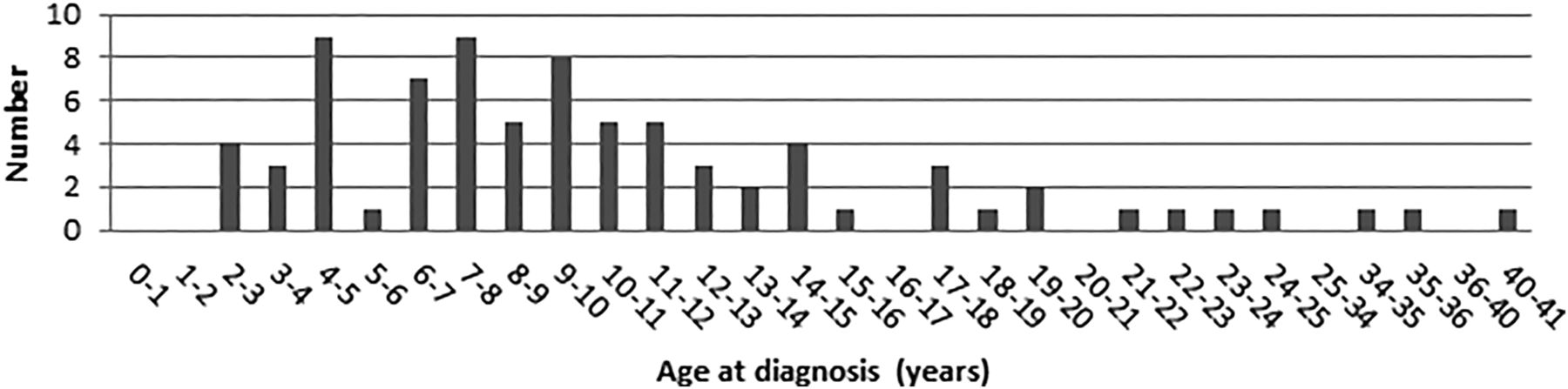
Age at diagnosis of brain tumours.

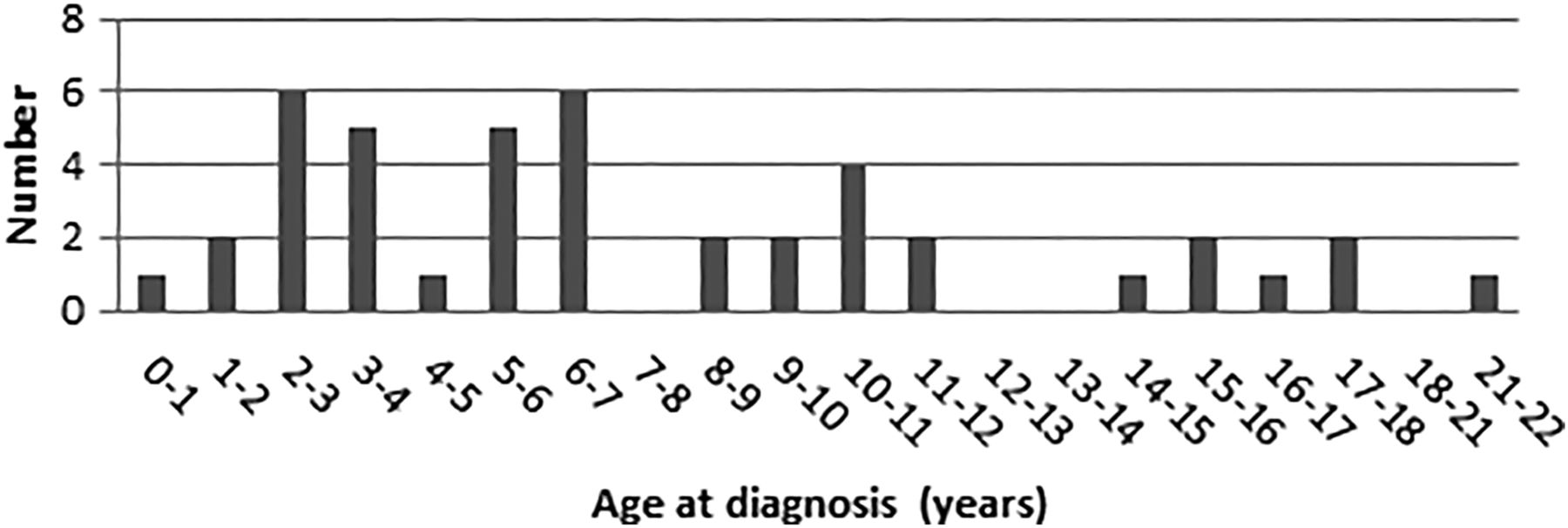
Age at diagnosis of lymphoma/leukaemia.
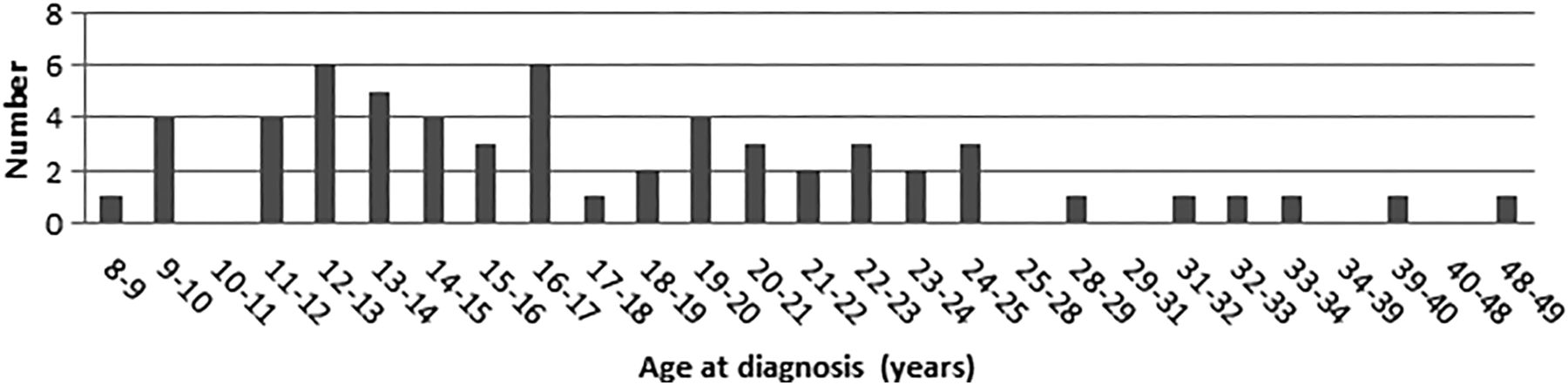
Age at diagnosis of colorectal cancer.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
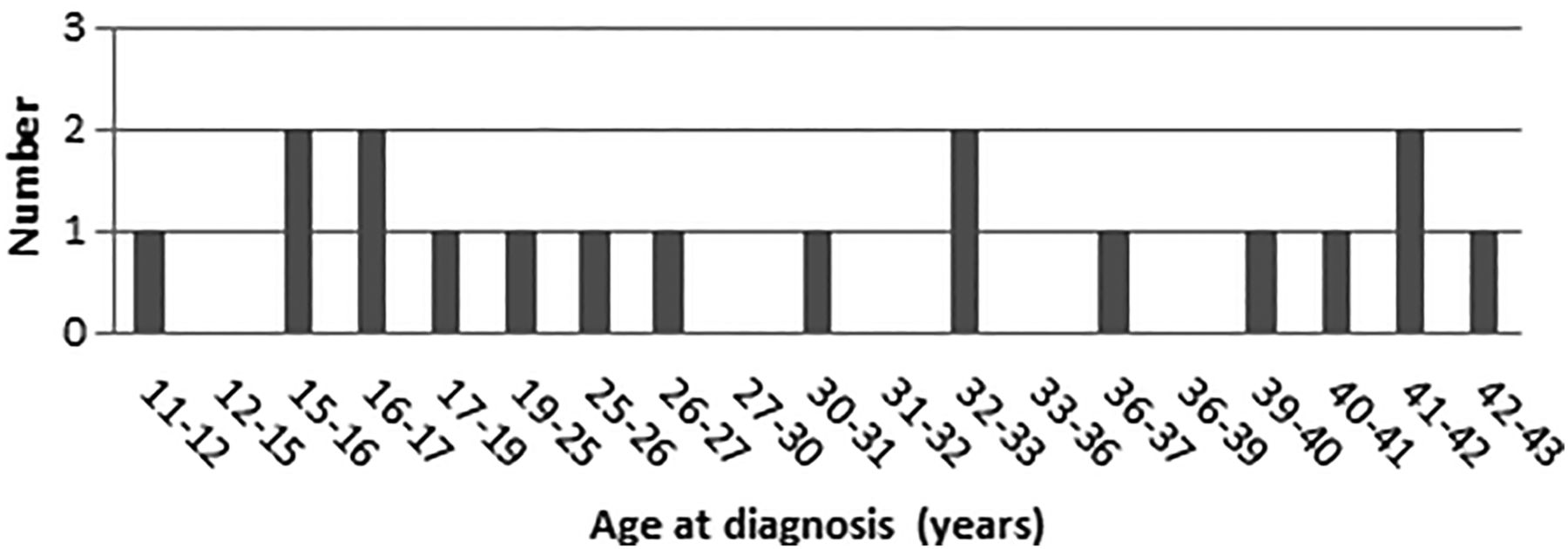
Age at diagnosis of small bowel cancer.
Clinical description and natural course of the most common cancers observed in CMMR-D
Brain tumours
Glioblastoma
Glioblastomas are tumours that originate from glia cells. According to the WHO grading system (2007), these tumours are classified as grade IV. The common symptoms and signs are related to raised intracranial pressure and include headache, nausea and/or vomiting and diplopia. Other symptoms include epileptic insults, unusual behaviour, and signs and symptoms caused by compression of parts of the brain and nerves.
The mean age at diagnosis in patients with CMMR-D is 9 years (range 2–40 years) (Wimmer et al, in preparation). When a patient is diagnosed due to clinical symptoms, half of all patients already have advanced disease. The diagnosis is suspected with MRI or CT scanning and confirmed by histological examination of a biopsy. Glioblastoma are characterised by rapid progression, and the usual treatment consists of surgical resection (if possible), followed by radiotherapy and/or chemotherapy depending on the age of the child. Due to diffuse growth, complete resection of the tumour can be achieved in less than 50% of cases7 and the reported 2-year survival in patients with sporadic glioblastoma is 30–50%.7 ,8 The prognosis mainly depends on the completeness of the surgical resection. If the tumour is completely resected, a median survival of 106 months has been reported.7 Glioblastoma is by far the most common cause of death in CMMR-D, it is not known whether the natural course and response to treatment of glioblastoma in CMMR-D differ from that of sporadic cases.
Other brain tumours
Other BTs reported in CMMR-D include medulloblastoma and (S)PNET. Medulloblastomas are located in the cerebellum and (S)PNET in other parts of the brain. These tumours derive from primitive neuroectodermal cells and are classified as WHO grade IV tumours. The mean age at diagnosis in patients with CMMR-D is 7 years (range 4–17 years). Frequently occurring signs and symptoms are related to increased intracranial pressure (nausea, headache, early morning vomiting) and are usually already present for some time prior to diagnosis but go unrecognised. In the general population, the estimated median delay from the first symptoms to diagnosis is around 2 months.9 At presentation, about 30% of tumours have metastasised via cerebrospinal fluid to other parts of the central nervous system. The diagnosis can be established using MRI and CT scanning. To evaluate whether the tumour has metastasised to the spinal cord, spinal MRI and a lumbar puncture are necessary. Treatment usually consists of surgical resection, radiotherapy (except in cases <3 years old) and chemotherapy. The prognosis is strongly dependent on the presence of metastases, and the prognosis of patients without metastases is relatively good (survival 75%) but neurocognitive consequences of brain radiotherapy may impair patient quality of life. Again, it is unknown whether the natural course of these tumours and the response to treatment in CMMR-D differ from that of sporadic cases.
Digestive tract cancers: colorectal cancer and small bowel cancer
CRC
It is generally accepted that most sporadic and hereditary colorectal cancers originate from adenomas. This also appears to hold for patients with CMMR-D, as colorectal adenomas are found in a third of all patients.2
Several studies have described digestive tract cancers in CMMR-D cases. In the 29 CMMR-D cases with CRC identified by Durno et al,10 the mean age at diagnosis was 16.4 years (range 8–28 years), 30 years younger than the typical age at diagnosis of CRC in LS. Information on the presence of adenomas was provided for a total of 18 out of the 29 patients with CRC. All but one had at least one adenoma, and 10 (55%) had multiple adenomas with numbers usually between 10 and 100. In addition, 11 out of 29 patients with CRC (38%) had multiple CRCs. There was no predilection for a specific site in the colorectum.
Herkert et al described four patients with CMMR-D with intestinal cancer and polyposis and biallelic PMS2 mutations. In addition, these authors identified all PMS2 CMMR-D cases with gastrointestinal manifestations published in the literature between 1980 and December 2009.11 They found 25 cases with gastrointestinal (small bowel and colorectal) polyps (mean age at diagnosis 17 years; range 7–46 years) and 42 cases with CRC (mean age at diagnosis 19; range 8–48 years). Full information on the colonic phenotype was available in 26 patients with CRC. Multiple adenomas (>10 adenomas) were found in 18 out of the 26 (70%) patients with CRC and multiple CRC in 38% of the cases.
Patients with CRC develop symptoms and signs, such as rectal blood loss, at a relatively late stage, and around 50% of the patients already have metastatic disease at diagnosis. The diagnosis of CRC is based on colonoscopy and confirmed by pathological examination of a tumour biopsy. The preferred treatment for CRC in young patients with LS is subtotal colectomy with ileorectal anastomosis (or proctocolectomy and ileal-pouch anal anastomosis in patients with rectal cancer). In patients with CMMR-D with multiple polyps (if there are too many to remove endoscopically and/or if they show high grade dysplasia) and patients with CRC, the treatment of choice would be colectomy with ileorectal anastomosis or proctocolectomy and construction of an ileal pouch-anal anastomosis.
Patients with stage III CRC (and advanced stage II CRC) receive chemotherapy, and those with rectal cancer are treated with radiotherapy. No specific side effects of radiotherapy have been reported in LS. However, the effectiveness and toxicity of chemotherapy and radiotherapy in CMMR-D is largely unknown (see below). Several studies have shown that there is an accelerated adenoma-carcinoma sequence in LS. Patients may develop CRC within 1–2 years after a normal colonoscopy. Our experience, supported by data from the literature on CMMR-D, indicates that adenoma and CRC development are also characterised by rapid progression. The 5-year survival for CRC in LS is approximately 50–60%.
Small bowel cancer
Cancer of the small bowel is also thought to originate from adenomas. Duodenal adenomas are found in 5% of CMMR-D cases (Wimmer et al, in preparation). In a series of 42 primary SBCs in LS, the mean age at diagnosis was 49 years (range 25–88 years), about 10–15 years younger than in sporadic SBC.12 Based on 18 SBCs in 12 patients, the mean age at diagnosis in CMMR-D is 25 years (11–42 years) (Wimmer et al, in preparation).
Herkert et al11 identified 25 cases with gastrointestinal (small bowel and colorectal) polyps (mean age at diagnosis 17 years; range 7–46 years) in the medical literature, and 11 with SBC (mean age at diagnosis 27; range 11–42 years).
About 50% of sporadic SBCs are located in the duodenum and 10–15% in the ileum, whereas in LS the cancers are more evenly distributed along the small bowel.12 The 11 PMS2 CMMR-D literature cases identified by Herkert et al11 developed 18 SBCs, including 8 duodenal cancers, 7 jejunal cancers and 3 ileal cancers. Three of the 11 patients developed multiple (up to 5) SBCs.5
In general, SBCs go unnoticed for a long period or manifest with only non-specific symptoms such as dull, cramping abdominal pain, abdominal distention and (faecal occult) blood loss. Obstruction is also a common presentation.
The diagnostic modalities used for assessing the presence of SBC are radiographic imaging (CT or MRI enteroclysis) and endoscopy (upper gastrointestinal (GI) endoscopy and ileocolonoscopy, for the detection of cancers located in the duodenum and ileum, respectively). There is also an increasing use of video capsule endoscopy (VCE) and double balloon enteroscopy. The effects of chemotherapy and radiotherapy are disappointing,13 and the treatment of choice is surgical resection. The 5-year survival rate of patients with resected tumours is around 50%. The natural history and prognosis of patients with CMMR-D with SBC are unknown.
Haematological cancers
Non-Hodgkin lymphoma
NHL is the most commonly occurring haematological cancer in CMMR-D (Wimmer et al, in preparation), with T cell NHL more frequently observed than B cell NHL. T cell NHL is usually located in the mediastinum, while B cell NHL has a mainly intra-abdominal location but is sometimes seen in the cervical region. Signs and symptoms vary depending on the type of lymphoma and the location, but may include coughing and respiratory distress (T cell NHL), obstruction of the bowel (B cell NHL), cervical lymphadenopathy, difficulty with swallowing (also seen in B cell NHL), and anaemia, tiredness and bruises in cases with bone marrow involvement.
Ages at diagnosis of sporadic NHL vary from 5 years to 12 years, according to histological subtypes.14 The median age at diagnosis of NHL in CMMR-D is 5 years (range 0.4–17 years), based on 31 cases (Wimmer et al, in preparation). In the general population, the median time to diagnosis, defined as the interval between the first signs and symptoms and diagnosis, is relatively brief (3.8 weeks).9 The techniques used to diagnose and stage NHL may include ultrasound of the abdomen, lymph nodes and testes, MRI or CT scanning, fluorodeoxyglucose-positron emission tomography (FDG-PET) scanning, biopsy of the tumour, bone marrow biopsy and lumbar puncture. Treatment for NHL consists mainly of intensive chemotherapy, with radiotherapy restricted to the small percentage of patients with overt central nervous system (CNS) disease at the time of diagnosis. The duration of treatment varies from a few weeks to 2 years, depending on the stage and the histological subtype. The prognosis is relatively good, with survival rates of 70–90%. The natural course and response to treatment of NHL associated with CMMR-D is unknown.
Acute leukaemia
The most common form of acute leukaemia in CMMR-D is acute lymphoblastic leukaemia (ALL). The mean age at diagnosis of ALL in CMMR-D is 6 years (range 2–21 years), based on nine cases (Wimmer et al, in preparation). The incidence of ALL peaks between 2 years and 5 years in non-CMMR-D. Children with ALL often present with signs and symptoms that reflect bone marrow infiltration and/or extramedullary disease including anaemia, thrombocytopenia, neutropenia and lymphadenopathy. Other presenting signs and symptoms are bone pain, fever, fatigue, bleeding and respiratory distress. The median time from the presentation of signs and symptoms to diagnosis is only 1–2 weeks.9 Tests required to classify ALL include immunotyping, cytogenetic studies and molecular studies to identify translocations. Lumbar puncture is performed to assess the involvement of the CNS. The diagnosis of ALL is confirmed by a bone marrow aspiration and biopsy. Although the treatment of ALL is primarily based on chemotherapy, the different forms of ALL require different approaches for optimal results. The prognosis of ALL depends on the clinical and laboratory features and the response to treatment. Overall, the cure rate of patients without CMMR-D with ALL is greater than 80%.
Considerations regarding the surveillance programme in patients with CMMR-D
Brain tumours
MRI scanning is the best screening method for the early detection of BT. Repeated CT scanning of the brain should be avoided because of the possible induction of tumours due to radiation.
As previously mentioned, glioblastomas usually show diffuse growth, meaning that discrimination between normal and tumour tissue may be impossible and the precise extent of the tumour may be difficult to assess. MRI scanning in young children is usually performed under general anaesthesia. MRI starting from birth is recommended by Durno et al.4 The youngest patients with CMMR-D diagnosed with glioblastoma were 2 years old. Therefore, we recommend commencing MRI scanning at the age of 2 years, and due to rapid progression, scanning at an interval of 6–12 months is probably needed. Whether early detection will lead to more complete surgical resections and improved survival is presently unknown.
Digestive tract cancer: CRC
Many studies have demonstrated that colonoscopy has the highest sensitivity and specificity and is thus the best tool for surveillance of the colon. In LS, small, flat and non-polypoid lesions are frequent and can easily be missed.15 ,16 We also observed mainly multiple non-polypoid lesions in a patient with CMMR-D. Therefore, it is recommended that chromoscopy be used in order to allow the detection and delineation of small, flat lesions. In children, colonoscopy is performed under general anaesthesia.
Based on experience with LS, surveillance of the colon is expected to be effective. Due to the assumed high progression rate from an adenoma to colorectal cancer, an intensive surveillance programme at annual intervals is probably needed. In patients with multiple adenomas, a shorter interval of 6 months is recommended. Because most cancers develop in the second decade of life, the programme can be started by the age of 8 years. In view of the large non-polypoid lesions often observed in CMMR-D, a paediatric gastroenterologist should perform the procedure together with an ‘adult’ gastroenterologist with experience of endoscopic mucosal resection of such tumours.
Digestive tract cancer: small bowel cancer
For the detection of duodenal cancers, an upper GI endoscopy can be performed (at the same time as colonoscopy). During colonoscopy, the terminal ileum should also be intubated for the identification of ileal cancer. CT scanning and MRI enteroclysis can be used for the detection of SBC located in the jejunum and remaining ileum but these modalities are too burdensome for surveillance purposes. A major disadvantage of regular CT scanning is the radiation burden. VCE is probably the best tool. Two studies in LS have shown that adenomas and SBC can be detected using VCE. In a French study by Saurin, tumours were detected in 10% of cases (one jejunal cancer, two adenomas).17 However, in a Dutch study on 200 LS mutation carriers, a tumour (one adenoma and one cancer) was identified in only 1.5% of cases (Haanstra et al submitted 2013). In this study, one patient developed a SBC 6 months after a normal VCE. The value of surveillance of the small bowel using VCE is therefore unknown. However, the high prevalence of such tumours (8%) in CMMD-R may justify the use of VCE. Because SBC below age 10 years is very rare, the surveillance programme can be started at the age of 10 years. Young children are generally able to swallow the capsule. An alternative is to place the capsule endoscopically during upper GI endoscopy.11
Non-Hodgkin lymphoma
Because sporadic T cell and B cell lymphomas have an excellent outcome, the benefit of early diagnosis is not obvious except for the avoidance of life threatening situations at diagnosis for patients with huge mediastinal masses. We currently have no information on the natural history of these lymphomas in CMMR-D, and while the natural course of the disease might differ from sporadic cases, sporadic cases usually present with rapidly growing tumours and clinical manifestations within a month prior to diagnosis. Thus, screening at less than 3-month intervals would probably be inefficient, whereas this frequency is probably too high for a screening for which we are not sure that it would improve the cure rate. A reasonable alternative is to perform clinical examinations, and optionally, abdominal ultrasound every 6 months. This strategy would probably be inefficient for early diagnosis but it might provide useful information on the natural history of this lymphoma in patients with CMMR-D.
Acute lymphocytic leukaemia/acute myeloid leukaemia
Signs and symptoms of acute leukaemia are apparent within a short period of time. As most patients with ALL have anaemia, thrombocytopenia with a normal or depressed white blood cells and lymphoblasts on peripheral smear, regular assessment of the blood count may be recommended but it is uncertain whether surveillance will lead to early detection and improvement of the prognosis.
Chemotherapy in Lynch syndrome and CMMR-D
About 15% of CRCs shows microsatellite instability (MSI), which is a marker of MMR deficiency. In sporadic CRC, MSI results from somatic MMR inactivation mainly due to epigenetic changes in the tumour, whereas in LS, MSI is caused by biallelic MMR gene inactivation due to a heterozygous germ line mutation and a somatic second-hit alteration in the other allele. Patients with CMMR-D show MSI due to a constitutional MMR defect caused by biallelic germ line MMR gene mutations.
A number of studies have evaluated the effectiveness of chemotherapy in the subgroup of CRC with MMR deficient versus MMR proficient tumours. Chemotherapy is the mainstay of treatment of many cancers in childhood. Due to the rarity of CMMR-D, very limited information on the optimal chemotherapy is available. Several studies have demonstrated that tumours with loss of MMR function are more frequently resistant to certain forms of chemotherapy.18 Another major concern is that some of these chemotherapeutic agents are mutagenic and may increase the risk of developing therapy-induced cancers, because patients with CMMR-D have a defect in DNA damage signalling and are unable to repair the accumulated somatic mutations.19
The effectiveness of chemotherapy in MMR-deficient CRC has recently been reviewed by Devaud and Gallinger.20 Evaluation of clinical trials performed in the 20th century that compared fluorouracil (5FU) with no treatment21 ,22 demonstrated that MSI-high (MSI-H) tumours (retrospectively analysed) were resistant to treatment with 5FU. These observations were confirmed by many in vitro studies.23 The resistance to 5FU of MMR-deficient cancer cells may be due to the incorporation of fluorouracil metabolites into DNA, although the sensitivity of MMR-deficient cell lines to this drug and other agents is likely to be dependent on the status of HSP110, a molecular chaperone that has been reported to be mutated in MMR-deficient tumours, sensitising MSI-tumour cells to a wide spectrum of anticancer agents including 5-FU24(Collura A, Gastroenterology, in press).
Although studies of MMR-deficient cell lines reported resistance to cisplatin and carboplatin, a good response to oxaliplatin was found and this was subsequently confirmed in clinical trials.13 ,25–28 Irinotecan also appears to be effective, similarly as in MMR-proficient tumours.29–32 Moreover, a recent study using cell lines showed a good response to irinotecan in combination with thymidine.33
Most of the above mentioned studies were performed in patients with somatic deficient MMR tumours. Less is known about the response in patients with LS and patients with CMMR-D with CRC.
All patients with CMMR-D with CRC and treated with chemotherapy described in the literature are shown in table 3.34–43 Most patients appear to show a response similar to the response in patients with sporadic CRC. However, prospective studies are needed to confirm this observation.
Response of colorectal cancer (CRC) to chemotherapy in CMMR-D
In 2007, Scott et al44 discussed the effectiveness of chemotherapy in patients with CMMR-D. Several cell line and mouse model studies showed that tumours are resistant to treatment with O6-methylating agents.18 One of these agents (temozolomide) is frequently used in the standard treatment of glioblastoma. This drug causes mutations in tumour DNA that cannot be repaired by patients with a loss of MMR function. Indeed, investigation of a clinical specimen from a patient treated with this drug showed an accumulation of somatic mutations (mutator phenotype).45 In vitro studies showed a similar effect for busulfan but not for chloroethylating agents such as cyclophosphamide and melphalan.18
MSI occurs in some tumours following therapy with thiopurines or cisplatin, suggesting that MMR deficiency is important in clinical resistance.46–48 In addition, MMR deficiency appears to be common in resistant acute myeloid leukemia and is a recurrent feature of secondary NHL occurring in allografted patients treated with azathioprine.49–51 A report on two patients with glioblastoma showed that they were resistant to treatment with temozolomide.52 Another study demonstrated loss of MSH6 expression in a subset of patients with glioblastoma resistant to temozolomide.53
All patients with CMMR-D known from literature with BT35 ,36 ,38 ,43 ,44 ,52 ,54–58 and lymphoma38 ,52 ,57 ,59–63 that were treated with chemotherapy are listed in tables 4 and 5. Most patients with T cell lymphomas showed a good response to chemotherapy. However, chemotherapy in patients with BT had a less favourable outcome. In particular, only one out of six patients treated with temozolomide and radiotherapy showed a partial response. In the other patients, the tumour was resistant to treatment.
Response of brain tumours (BTs) to chemotherapy in CMMR-D
Response of NHL to chemotherapy in CMMR-D
Whether temozolomide or other drugs such as cisplatin and busulfan are contraindicated in CMMR-D is currently controversial and requires further studies. As stated by Scott et al,44 early detection of tumours may allow considerations about the most effective chemotherapeutic regimen.
Discussion
For many types of cancers, diagnosis at the earliest possible (preferably preclinical) stage results in much more effective treatment and an improved prognosis. This may also be the case for CMMR-D. Based on the available knowledge and the discussions at the workshop, the European consortium proposed a surveillance protocol as summarised in table 6. The surveillance for CRC is the only aspect that largely complies with the WHO criteria for screening, although it is unknown whether colonoscopic surveillance in CMMR-D is as effective as in LS. The value of all other suggested screening protocols is unknown. In particular, it is questionable whether surveillance for ALL and NHL improves the already favourable outcome for patients with these tumours. A randomised controlled trial is needed to assess whether surveillance can improve the prognosis. However, the question then arises whether it is ethically justified not to offer surveillance in view of the high mortality without surveillance. As most parents/patients would probably choose to participate in a surveillance programme, performing a trial would be difficult. The best approach may be to discuss the advantages and disadvantages of surveillance and to make a joint decision (table 7).
Surveillance protocol for patients with CMMR-D proposed by the European consortium
Pros and cons of participating in a surveillance programme
A general recommendation is that parents (or adult patients) should contact their doctor if the child (adult patient) develops unusual signs or symptoms. This is especially important in the case of BT, in view of the rather long time to diagnosis.
The programme recommended by the consortium aims to detect the most common cancers. However, a variety of other tumours also occur in CMMR-D (table 2). A prospective randomised trial may be performed to test whether a once yearly (rapid) whole body MRI adds some efficacy to our screening programme.
As already mentioned above, it should be emphasised that the distribution of ages at diagnosis of the various cancers is biased by collection of published cases. In fact, ascertainment bias may be a major issue in all that we know about this condition. As further study is done, less severe cases may possible be seen more frequently.
Nevertheless, the high risk of developing a wide variety of cancers, the high risk of developing multiple cancers, sometimes synchronously, the occurrence of these cancers at a young age and the need for intensive multimodal treatment in the case of cancer, imposes an enormous burden on parents and patients. A recent evaluation of psychological distress in individuals predisposed to develop cancer showed increased levels of distress in adults at risk of developing multiple tumours.64 Nothing is known about the psychological distress in patients with CMMR-D and their parents. Therefore, doctors involved in the care of these patients should offer the support of a psychologist.1 In addition, the clinical geneticist plays an important role by organising presymptomatic testing of other family members for CMMR-D or LS, and through discussion of the option of prenatal or preimplementation genetic diagnosis.
Individuals at risk for multiple cancers usually see several doctors including paediatric oncologists, neurologists, (neuro)surgeons, gastroenterologists and haematologists. It is important that one of these specialists coordinates the surveillance examinations and is available for the patients if they have questions.
A prerequisite for participation in a surveillance programme is prospective collection of all results of surveillance, including the response to treatment. With this aim in mind, the European consortium established an EU-CMMR-D database. This European registry will allow the (cost)effectiveness of the surveillance programme to be evaluated and will allow the sensitivity of these tumours to chemotherapy to be assessed.
Conclusion
In an era of high-throughput sequencing technologies, an increasing number of patients with CMMR-D are being identified. Most patients die from cancers in early childhood. The only way to improve the prognosis is surveillance for these cancers. Surveillance for CRC, and possibly SBC, might be effective; however, the value of surveillance and early detection of BT and NHL/leukaemia is unknown. The best approach is to discuss the advantages and disadvantages of surveillance. When patients and parents decide to participate, outcomes should be collected and evaluated within the EU registry.
Acknowledgments
The authors thank the following colleagues for their contribution during the discussions at the meeting in Paris on 8 June 2013: Felipe Andreiuolo, Centre National de la Recherche Scientifique, Gustave Roussy Cancer Insitute, Villejuif, France; Julie Tinat, Department of Genetics, Faculty of Medicine, University of Rouen, Rouen, France; Marina Dimaria, Department of Genetics, Gustave Roussy Cancer Institute, Villejuif, France; Ada Collura, Basic and translational research, Hospital St Antoine, Paris, France; Cécile Charpy, Department of Pathology, Gustave Roussy Cancer Institue, Villejuif, France; Olivier Buhard, Basic and translational research, Hospital St Antoine, Paris; Sarah Bodo, Basic and translational research, Hospital St Antoine, Paris.
References
Footnotes
-
Contributors The guidelines have been discussed during the workshop in Paris. All authors were involved in this discussion. The manuscript was written by HFAV and ZG with specific contributions from KW and LB
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.