Article Text

Abstract
Background Split hand/split foot malformation (SHFM) type 1 is characterised by missing central digital rays with clefts of the hands and/or feet, which was linked to chromosome 7q21.3. While double knockout of Dlx5 and Dlx6 resulted in limb defects in mice, the majority of patients with SHFM1 had only heterozygous chromosomal abnormalities.
Objective To investigate the clinical and molecular features of a large family with SHFM1.
Methods Blood samples of family members were investigated by linkage analysis, array comparative genomic hybridisation, exome sequencing and PCR-Sanger sequencing. Cultures from bone specimens obtained from the proband and an unrelated unaffected individual were established and subjected to quantitative real-time PCR, reverse-transcribed PCR, Western blot and imprinting analysis.
Results We report a large pedigree of SHFM1 with 10 members having a heterozygous 103 kb deletion, the smallest one ever reported to be associated with SHFM1. Of these 10, two had no limb anomalies, making a penetrance of 80%. The deletion encompassed exons 15 and 17 of DYNC1I1, which are known enhancers of two downstream genes, DLX5 and DLX6. Surprisingly, DLX5 and DLX6 RNA and proteins in our proband's cultured osteoblasts, instead of 50% decrease, were absent. Allelic expression studies in cultured osteoblasts of the unaffected individual showed that DSS1, DLX6 and DLX5 expressed only paternal alleles. These lines of evidence indicate that DSS1, DLX6 and DLX5 were maternally imprinted in osteoblasts.
Conclusions SHFM1 in our family is caused by a heterozygous paternal deletion of enhancers of the osteoblast-specific maternally imprinted DLX6 and DLX5 genes, leading to the absence of their proteins.
- Imprinting
- Molecular genetics
- Developmental
Statistics from Altmetric.com
Introduction
Split hand/split foot malformation (SHFM) is one of the most complex limb abnormalities, characterised by missing central (second, third and/or fourth) digital rays with clefts of the hands and/or feet and fusion of remaining digits.1 The prevalence of SHFM has been reported to be 0.60/10 000 in Caucasians.2 SHFM can be presented in either an isolated form or in combination with additional anomalies.3
SHFM disorders are genetically heterogeneous with multiple loci and different inheritance patterns. For the isolated form, nine different genetic loci have been mapped and two genes have been identified: SHFM1 (MIM 183600) at chromosome 7q21.3, SHFM2 (MIM 313350) at Xq26, SHFM3 (MIM 246560) at 10q24, SHFM4 (MIM605289) due to mutations in P63 at 3q27, SHFM5 (MIM606708) at 2q31 and SHFM6 (MIM225300) caused by mutations in WNT10B at 12q13. In addition, three more loci on 8q21.1–q22.3,4 4q32–4q355 and 6q16–6q225 were reported to link to SHFM.
The 7q21.3 region associated with SHFM1 includes the candidate genes DSS1, DLX5 and DLX6, which are important in limb development.6 ,7 Knocking out both alleles of either Dlx5 or Dlx6 was lethal in mice and showed craniofacial defects, but did not lead to limb abnormalities. To manifest SHFM phenotype, both alleles of Dlx5 and Dlx6 were needed to be knocked out.8 ,9 In humans, the majority of patients with SHFM1 were heterozygous for chromosomal structural abnormalities. Only two families with limb defects were found to have point mutations in DLX5. The first was a consanguineous family with two siblings having SHFM and hearing loss. Both were found to have a homozygous missense mutation in DLX5, c.A533C (p.Q178P).10 The second family had two affected members, a mother and her son, with split feet and triphalangeal thumbs. Both had a heterozygous missense mutation in DLX5, c.G558T (p.Q186H).11 All other patients associated with the SHFM1 locus were caused by deletions or complex chromosomal rearrangements.
Studies in both mice and zebrafish have identified novel tissue-specific enhancers located within the coding regions of Dync1i1 (dynein, cytoplasmic 1, intermediate chain 1) that regulate Dlx5 and/or Dlx6.7 A recent study identifying a 106 kb deletion encompassing exons 11–18 of the SLC25A13 gene and exons 14–17 of the DYNC1I1 gene in patients with SHFM1 has confirmed a key role of DYNC1I1 exonic enhancers in normal limb formation in humans.12
Here, we describe a large pedigree with SHFM1. A heterozygous 103 kb deletion, the smallest ever reported, encompassing parts of DYNC1I1 and SLC25A13, was identified. Surprisingly, DLX5 and DLX6 proteins in our proband's cultured osteoblasts were absent. Allelic expression studies in cultured osteoblasts of an unaffected individual showed that DSS1, DLX6 and DLX5 were maternally imprinted in osteoblasts. These findings indicate that SHFM1 in our family is caused by a heterozygous paternal deletion of enhancers of the osteoblast-specific maternally imprinted DLX6 and DLX5 genes, leading to the absence of their proteins.
Patients and methods
Linkage analysis
After written informed consent was obtained, genomic DNA was extracted from peripheral blood leucocytes using commercially available kits (Qiagen, Valencia, California, USA). We performed linkage analysis in 14 family members by genotyping microsatellite markers at eight autosomal SHFM loci. This included SHFM1 on chromosome 7q21 (D7S657 and D7S515), SHFM3 on chromosome 10q24 (D10S192 and D10S597), SHFM4 on chromosome 3q28 (D3S1601 and D3S1580), SHFM5 on chromosome 2q31 (D2S335 and D2S364), SHFM6 on chromosome 12q13 (D12S368) and loci on chromosome 4q32–4q35 (D4S413, D4S1597 and D4S1539), 6q16–6q22 (D6S434, D6S287 and D6S262) and 8q21.11–q22.3 (D8S270 and D8S1784). The details of the primers were obtained from Marshfieldmap (http://research.marshfieldclinic.org/genetics/home/index.asp). We typed all fluorescently labelled PCR products on an ABI Prism 3100 genetic analyser (Applied Biosystems, Foster City, California, USA) with GeneMapper software (Applied Biosystems). The MLINK program (available from http://linkagerockefeller.edu/.rockefeller.edu/) was used to calculate the two-point linkage analysis with the model of autosomal-dominant inheritance with a high penetrance.
Copy number analysis by array comparative genomic hybridisation
Genomic DNA of the proband and a normal Thai control were sent to Macrogen (Seoul, South Korea) to determine copy number variations. DNA was independently labelled with fluorescent dyes, co-hybridised to specific sequences on chromosome 7 using NimbleGen 385K chip (cat. no. B3738001-00-01; Roche NimbleGen Systems, Madison, Wisconsin, USA) and scanned using a 2 µm scanner. Log2-ratio values of the probe signal intensities (Cy3/Cy5) were calculated and plotted versus genomic position using Roche NimbleGen NimbleScan software. Data were displayed in Roche NimbleGenSignalMap software.
Breakpoint identification
To define the breakpoints, we performed PCR with multiple pairs of primers flanking the deletion including FLANK-F: 5′-CCGCCCATACAACCTCATTT-3′ and FLANK-R: 5′-GATGT CTGAAGTCAGCAACC-3′, followed by Sanger sequencing.
Segregation analysis and screening for the deletion in ethnic matched controls
To determine SHFM1 genotypes in all 19 available family members, we used a duplex PCR strategy. Two pairs of primers, FLANK-F and FLANK-R to amplify the 2245 bp deleted allele on chromosome 7q21 and 5′-CTTGTGTACTTTGGCTTTC ATTCAC-3′ and 5′- GCTCTATATTTACTGCAGCACAGAAC-3′ to amplify a 1188 bp control allele of KMT2C, were used for PCR amplification. PCR products were resolved on a 1.2% agarose gel. This pair of primers was also used to analyse 150 unrelated unaffected Thai controls.
Determination of point mutations in candidate genes
PCR amplification of all coding regions of the DYNC1I1, DSS1, DLX6 and DLX5 genes was carried out using primers and annealing temperatures in online supplementary table S1. PCR products were treated with ExoSAP-IT (USP Corporation, Cleveland, Ohio, USA) and sent for direct sequencing at Macrogen. Sequence data were analysed using Sequencher (V.5.0; Gene Codes Corporation, Ann Arbor, Michigan, USA).
Exome sequencing
Exome sequencing using high-quality genomic DNA from peripheral blood of the proband (IV-30) was performed by Macrogen. DNA was captured on the TruSeqExome Enrichment Kit (Illumina) and subsequently sequenced on the Hiseq2000 instrument. Sequence reads were mapped against UCSC hg19 using BWA software (http://bio-bwa.sourceforge.net/). The SNPs and Indels were detected by SAMTOOLS (http://samtools.sourceforge.net/) and annotated by dbSNP&1000G. Then we sought for pathogenic mutations in the 26 candidate genes in the 9 SHFM loci, including BHLHA9, BTRC, CHD3, DACTYLIN, DLX1, DLX2, DLX5, DLX6, EVX2, EPS15L1, FGF8, FGF13, HOXD2, HOXD5, HOXD6, HOXD7, HOXD9, HOXD10, HOXD11, HOXD12, HOXD13, LBX1, PBXW4, POLL, TP63 and WNT10B.3 ,13
Human bone culture
Bone tissues were taken from the proband (IV-30) during an operation to close the cleft on his right hand. We obtained a bone chip including cortical and cancellous bones from his right third metacarpal. A bone chip of an unrelated unaffected individual undergoing surgery after an accident was also obtained. Cells were seeded in a T-25 cell culture flask, cultured in DMF-12 (Gibco) supplemented with 20% fetal bovine serum, 1% penicillin–streptomycin and incubated in a humidified atmosphere with 5% CO2 at 37°C. Passages 3–5 were used for experiments. Osteoblast differentiation was induced by the addition of 50 μg/mL ascorbic acid and 10 nM dexamethasone.
RNA expression of cultured osteoblasts
After three passages, total RNA was extracted from bone cultures using a QIAamp RNA blood mini kit (Qiagen). Reverse transcription was performed using ImProm-IITm RT (Promega, Madison, Wisconsin, USA). RNA levels of DYNC1I1, DSS1, DLX6 and DLX5 were quantified by quantitative real-time (qRT) analysis using TaqMan probes of the StepOnePlus Real-time PCR system (Applied Biosystems) (see online supplementary table S2). For DYNC1I1, we performed qRT using two pairs of primers; one for amplifying exons 11–12, 5′ to the deletion, and the other for amplifying exons 16 and 17, which were within the deletion. The probes were run in triplicate two times in separate tubes. Relative expression analysis was calculated in terms of ΔΔCt normalised to GAPDH transcription levels. We also determined the expression ratio of the DYNC1I1, DSS1, DLX6 and DLX5 in each bone culture sample by PCR using complementary DNA (cDNA) as template. The PCR products were separated on a 2% agarose gel to analyse gene transcripts. Sequences of primers are available in online supplementary table S3.
Protein expression of cultured osteoblasts
The antibodies including a monoclonal mouse anti-DYNC1I1 (catalogue #ab23905, Abcam, Cambridge, UK), a monoclonal rabbit anti-DLX6 (catalogue #ab137079, Abcam), a polyclonal rabbit anti-DLX5 (catalogue #ab109737, Abcam), a monoclonal rabbit anti-alpha tubulin (catalogue #ab52866, Abcam) and a polyclonal rabbit anti-DSS1 (catalogue #1, Proteintech, Chicago, Illinois, USA) were obtained.
Protein was extracted from bone cultures using ice-cold RIPA lysis buffer with Halt protease inhibitor cocktail (Pierce, Rockford, Illinois, USA) and protease inhibitor cocktail (Sigma Aldrich, Sigma, Missouri, USA). The total soluble protein concentrations were determined using the bicinchoninic acid protein assay reagent (Pierce). Sodium dodecyl sulfate (SDS)-containing buffer was added to supernatant and the sample was denatured by heating for 7 min before SDS-polyacrylamide gel electrophoresis (PAGE) loading. After size separation by 4%–15% SDS-PAGE, proteins were transferred onto polyvinylidene difluoride membranes by iBot Gel Transfer System (Invitrogen).
The membrane was stripped and reprobed with 1:1000 antialpha tubulin (Abcam) and 1:1500 goat anti-rabbit IgG HRP: sc-2030 (Santa Cruz Biotechnology, Santa Cruz, California, USA).
Imprinting analysis
We obtained a blood sample and a bone chip from the right femur of an unaffected individual during an operation for his fracture. Blood samples from his parents were also obtained. Genomic DNA was extracted from their blood samples. Osteoblasts were cultured from the bone chip. RNA was extracted from the cultured osteoblasts and reversed transcribed to cDNA. PCR using genomic DNA of this individual followed by direct sequencing of the DSS1, DLX5 and DLX6 entire coding, 5′UTR and 3′UTR regions was performed.
SNPs were identified within exons of DSS1, DLX6 and DLX5 using the National Center for Biotechnology Information SNP database (http://www.ncbi.nlm.nih.gov/SNP/). Primer sequences used for allele-specific expression analyses for DSS1, DLX6 and DLX5 are available in online supplementary table S3. We incubated the total RNA with or without RT, including an oligo (dT)15 primer to confirm that no PCR amplification signals could be detected in the absence of RT. The regions of the genomic DNA and cDNA containing SNPs were PCR-amplified using HotStarTaq DNA polymerase (Qiagen) and primers (see online supplementary table S3). The PCR products were subjected to Sanger sequencing. Allelic expression was determined by comparing sequences of the child's cDNA and his parents’ genomic DNA.
Results
Clinical studies
We investigated 19 members from a large Thai family including 8 affected and 11 unaffected with SHFM (figure 2A, see online supplementary figure S1). None reported fractures or had ear abnormalities. Physical examination revealed that the spectrum of clinical manifestations was broad (figure 1, see online supplementary table S1 and figure S2), ranging from the presence of cutaneous syndactyly between the second and third digits of right hand as in III-4 (see online supplementary figure S2) to bilateral split hands and feet in II-11 (figure 1).

Clinical and radiological features of hands and feet of patients.
Linkage analysis
We first performed linkage analysis in 14 individuals on eight known autosomal loci (SHFM1, SHFM3-6, 4q32–4q35, 6q16–6q22 and 8q21.11-q22.3). We were able to exclude linkage to all loci except the SHFM1 locus on chromosome 7q21. Using markers D7S657 and D7S515, the maximum two-point logarithm of odds score was 2.41 at zero recombination (figure 2A).

(A) Pedigree of the family and genotype of microsatellite markers in the region of split hand/split foot malformation-1 on chromosome 7q21.3. Affected individuals are represented by filled symbols and unaffected individuals by open symbols. The boxes defined by microsatellite markers D7S657 and D7S515 cosegregate with the disease. (B) PCR amplification of the 103 kb deletion allele and KMT2C, used as a control gene. Lane 1 contains a DNA size marker (M) with an arrow head indicating a size of 1000 bp; lanes 2–19 represent the respective family members. The upper bands present in II-2, III-4, IV-9, IV-10, II-7, III-22, IV-30, II-11, III-29 and III-30 are the amplified products of the deletion allele (2245 bp). The lower 1188 bp bands present in all family members are a part of the KMT2C gene, used as a PCR control.
Copy number analysis and breakpoint identification
Using a targeted 385K chromosome 7-specific microarray, we identified an approximately 100 kb deletion on 7q21 (figure 3A). Sanger sequencing of PCR products spanning the breakpoints revealed a 103 763 bp deletion, g.95694099_95797866delins TCATC (figure 3B), encompassing exons 14–17 of the DYNC1I1 gene and exons 13–18 of the SLC25A13 gene. The deletion did not encompass an otic vesicle enhancer that was found to be located in SLC25A.

(A) Previously reported chromosome 7 deletions in split hand/split foot malformation-1 and (B) sequencing chromatogram of the breakpoints of our proband showing the distal and proximal breakpoints with a 5 bp insertion.
Segregation analysis and screening for the deletion in ethnic matched controls
All 19 available family members were genotyped. We found that the deletion was present in all eight patients and in two unaffected individuals (III-30 and IV-9) (figure 2B), but absent in 150 unaffected control individuals (300 chromosomes).
Exclusion of point mutations
To be certain that no point mutations in the coding regions of DYNC1I1, DSS1 DLX6 and DLX5 contributed to or modified the phenotype, we PCR-sequenced the entire coding regions of these four genes in the proband and found no mutations.
Exome sequencing
To exclude other pathogenic mutations in genes within the 9 linked loci of SHFM that might affect the phenotype of our proband, exome sequencing was performed.
Exome sequencing generated 5.83 Gb of sequence data, with 71.4% of target regions covered at least 10 times. No pathogenic mutations in the 26 genes linked to the SHFM loci were identified.
Osteoblast culture and determination of DLX5 and DLX6 expression levels
As enhancers of DLX5 and DLX6 reside in exons 15 and 17 of DYNC1I1, we hypothesised that the deletion encompassing exons 14–17 of DYNC1I1 could alter the expression of nearby genes leading to the SHFM phenotype. We used qRT PCR, PCR using cDNA as template and Western blot analysis to determine RNA and protein levels of DYNC1I1, DSS1, DLX5 and DLX6 in the proband's cultured osteoblasts. The RNA level of DYNC1I1 using primers amplifying exons 11–12, which were 5′ to the deletion, was 46% of the control. This was statistically significantly higher than 32% of the control when primers amplifying exons 16–17, which were within the deletion, were used (see online supplementary figure S3). As expected, the protein levels of DYNC1I1 were approximately half compared with the control. RNA and protein levels of DSS1 were similar to those of controls (figure 4). Surprisingly, RNA and protein of DLX5 and DLX6 were absent (figure 4).
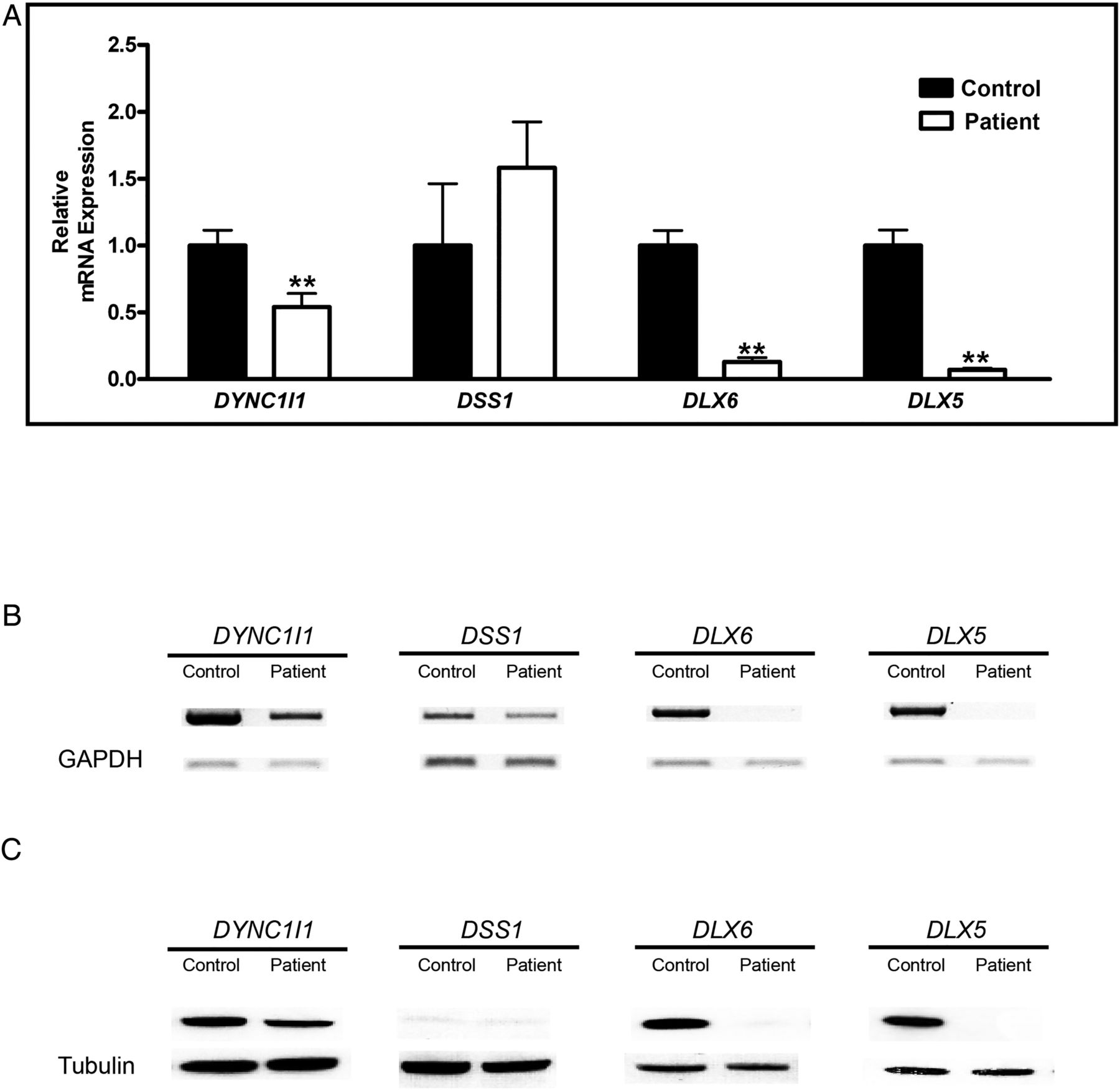
Expression of DYNC1I1, DSS1, DLX6 and DLX5 in cultured osteoblasts of our proband and an unaffected individual. (A) RT-PCR analysis: graphical representation of the relative RNA expression. (B) Semi-quantitative assay. Total RNA was used for cDNA synthesis; glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control. (C) Western blot analysis.
Imprinting analysis
Since the DLX5 and DLX6 of the proband who harboured a heterozygous paternal deletion were not expressed, we hypothesised that both were maternally imprinted genes. We studied the imprinting phenomenon of these genes in osteoblasts of an unrelated unaffected individual. We first identified heterozygous variants whose parental origin could be determined in DSS1, DLX5 and DLX6. At the DSS1 7:96317968 position (hg 19), the individual, his father and his mother were CT, CC and CT, respectively. At the DLX5 rs 73708843, the individual, his father and his mother were CT, CC and CT, respectively. At the DLX6 rs 2272280, the individual, his father and his mother were CT, TT and CC, respectively.
We then PCR-sequenced this unaffected individual's cDNA from cultured osteoblasts, which showed that only the C allele of DSS1, the C allele of DLX5 and the T allele of DLX6 were expressed. All were paternal alleles (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Imprinting analysis of the DSS1, DLX6 and DLX5 genes in cultured osteoblasts of an unaffected individual. The three upper panels used genomic DNA of leucocytes as template for PCR, while the lowest panel used complementary DNA, reversed-transcribed from RNA extracted from an unaffected individual's cultured osteoblasts.
Discussion
We identified a large Thai pedigree with 10 members having a 103 763 bp deletion encompassing the exons 15 and 17 of DYNC1I1. The trait exhibited a great range of severity. Of these 10, two members (III-30 and IV-9) had absolutely no abnormalities, making a penetrance rate of 80%. Among the remaining eight members who exhibited limb abnormalities, the severity ranged from mild cutaneous unilateral syndactyly of the second and third digits (III-4), to severe involvement of both hands and feet (II-11). Besides non-penetrance and variable expressivity, we observed an unusual phenotype. The proband's father (III-22) showed a central polydactyly of his right hand (see online supplementary figure S2). Unfortunately, we do not have pictures or radiographs of his right hand before surgery to remove the extra digit. No polydactyly has previously been reported in SHFM1.
We then performed experiments to determine the molecular pathology in our family with SHFM. Because eight autosomal loci have been linked to SHFM, we started with linkage analysis using 2–3 microsatellite markers for each locus. Seven loci were excluded, leaving only the SHFM1 locus. We subsequently went on to examine whether there was a deletion in this locus. Using array comparative genomic hybridisation followed by PCR and direct sequencing, we were able to identify the deletion and its breakpoints causing SHFM in this family. Previously the SHFM1 critical region was around 888 000 bp14 and very recently was narrowed down to 105 935 bp.12 Here, we report the smallest 103 763 bp deletion encompassing exons 14–17 of the DYNC1I1 gene and exons 13–18 of the SLC25A13 gene. The deletion was found in all eight affected and two unaffected members. PCR and direct sequencing of the entire coding regions of the DYNC1I1, DSS1, DLX6 and DLX5 genes showed no mutations. Exome sequencing revealed no mutations of genes in the linked SHFM loci. We, therefore, concluded that the molecular pathology, causing SHFM in our family, was this 103 763 bp deletion.
Since DYNC1I1 is not expressed in limbs during embryogenesis15 and homozygous SLC25A13 mutations cause citrin deficiency (MIM 603859),16 both genes are unlikely to be responsible for SHFM1. As previously reported in zebrafish, mice and humans, the exons 15 and 17 of DYNC1I1 also act as enhancers of DLX5/6 expression.7 ,17 Both genes express in limbs during mouse embryonic day 12.5.8 ,9 ,18 ,19 We hypothesised that haploinsufficiency of DLX5 and DLX6 caused SHFM1. After obtaining a bone chip of the proband, we cultured osteoblasts and determined expressions of DYNC1I1, DSS1, DLX5 and DLX6. RNA and protein levels of DYNC1I1 were approximately half of the control. Although DSS1 protein was technically difficult to detect,20 we showed that RNA and protein levels of DSS1 were similar to those of the control. Surprisingly, RNA and protein levels of DLX5 and DLX6 of the proband's cultured osteoblasts were absent, instead of being half of those of the control. This is consistent with findings in knockout mouse experiments. Only homozygous dlx5/dlx6 double-knockout mice showed a split hand/foot type I phenocopy, while heterozygous dlx5/dlx6 double-knockout mice showed no obvious limb defects.8 ,9
Since only one allele was deleted but no protein was present, we hypothesised that DLX5 and DLX6 were imprinted genes. Moreover, the pedigree revealed that all patients inherited the deleted allele from their father. Thus, these findings suggest that DLX5 and DLX6 are maternally imprinted genes. Interestingly, DLX5 was previously shown to be expressed only from the maternal allele in brain and peripheral blood cells.21 ,22 We studied allelic expression of DSS1, DLX5 and DLX6 in cultured osteoblasts obtained from the right femur of an unaffected individual. We found that all three genes were expressed only from the paternal allele, confirming that all the three genes are maternally imprinted in osteoblasts.
Since the DSS1 RNA and protein levels of our proband were not different from those of the control, this suggested that DNA in the deleted region did not regulate the expression of DSS1. On the contrary, the RNA and protein of DLX5 and DLX6 of our proband were absent, suggesting that enhancers of these two genes were in the deleted region. This confirms the role of DYNC1I1 exonic enhancers in normal limb formation in humans.
There were seven previously reported patients in five unrelated pedigrees with SHFM1 who had chromosomal deletions with known parental origins.12 ,23–26 Six inherited the deletions from their fathers. There was only one patient with a split left hand who inherited the deletion from his non-penetrant mother. Further studies are required to elucidate this unexpected finding.
In summary, we reported the smallest deletion associated with SHFM1, confirmed the role of DYNC1I1 exonic enhancers in development of human limb buds and found for the first time that DSS1, DLX5 and DLX6 were maternally imprinted genes in osteoblasts. SHFM1 in our family is caused by a heterozygous paternal deletion of the enhancers of the osteoblast-specific maternally imprinted DLX6 and DLX5 genes, leading to the absence of their proteins.
Acknowledgments
We thank the family members for participating in this study, Dr Noppachart Limpaphayom and Dr Kawee Pataradool for collecting bone samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors SR performed molecular genetics and functional studies and drafted the manuscript. ST performed mutation analysis. CS performed quantitative real-time PCR analysis. PK coordinated sample collection and collected the clinical data. KS and VS designed, supervised the study and participated in the draft of the manuscript. All authors read and approved the final manuscript.
-
Funding This study was supported by the Royal Golden Jubilee Ph.D. Program to SR (grant no. PHD/0071/2554), the Ratchadapiseksomphot Endowment Fund of Chulalongkorn University (RES560530177-HR) and the Thailand Research Fund (RTA5680003).
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Data from this study that do not pertain to individual patients are freely available, in accordance with the principles of the funding agency, and can be obtained by contacting the authors.