Article Text

Abstract
Background Infants with Noonan syndrome (NS) are predisposed to developing juvenile myelomonocytic leukaemia (JMML) or JMML-like myeloproliferative disorders (MPD). Whereas sporadic JMML is known to be aggressive, JMML occurring in patients with NS is often considered as benign and transitory. However, little information is available regarding the occurrence and characteristics of JMML in NS.
Methods and results Within a large prospective cohort of 641 patients with a germline PTPN11 mutation, we identified MPD features in 36 (5.6%) patients, including 20 patients (3%) who fully met the consensus diagnostic criteria for JMML. Sixty percent of the latter (12/20) had severe neonatal manifestations, and 10/20 died in the first month of life. Almost all (11/12) patients with severe neonatal JMML were males. Two females who survived MPD/JMML subsequently developed another malignancy during childhood. Although the risk of developing MPD/JMML could not be fully predicted by the underlying PTPN11 mutation, some germline PTPN11 mutations were preferentially associated with myeloproliferation: 10/48 patients with NS (20.8%) with a mutation in codon Asp61 developed MPD/JMML in infancy. Patients with a p.Thr73Ile mutation also had more chances of developing MPD/JMML but with a milder clinical course. SNP array and whole exome sequencing in paired tumoral and constitutional samples identified no second acquired somatic mutation to explain the occurrence of myeloproliferation.
Conclusions JMML represents the first cause of death in PTPN11-associated NS. Few patients have been reported so far, suggesting that JMML may sometimes be overlooked due to early death, comorbidities or lack of confirmatory tests.
- Haematology (Incl Blood Transfusion)
- Molecular Genetics
- Paediatric Oncology
Statistics from Altmetric.com
Introduction
Noonan syndrome (NS, OMIM 163950) is a relatively common (1/2000 births) developmental disorder characterised by reduced postnatal growth, congenital heart defects and cardiomyopathy, variable cognitive deficits, and distinctive facial dysmorphism.1 NS patients have an increased risk of developing several types of childhood malignancies,2 ,3 including myeloproliferative disorders (MPD) resembling juvenile myelomonocytic leukaemia (JMML).4
MPDs are clonal haematopoietic diseases associating variable degrees of myeloproliferation of different cell lineages.5 JMML is a rare and aggressive myelodysplastic and myeloproliferative neoplasm of early childhood, associated with excessive monocytic and macrophagic proliferation.6 ,7 Patients typically present with splenomegaly, monocytosis, anaemia, thrombocytopenia and elevated fetal haemoglobin (HbF). Myelograms reveal less than 20% of myeloblasts, and varying degrees of aberrant myelopoiesis or megakaryopoiesis.8 A hallmark of JMML is the hypersensitivity of myeloid progenitors to granulocyte-macrophage colony-stimulating factor (GM-CSF).9 Sporadic JMML is usually aggressive and has a poor prognosis. The only curative treatment is allogeneic bone marrow transplantation, with a relapse rate of 30–40%. By contrast, previous reports have suggested that NS-related JMML is often benign and resolves spontaneously.4 ,10–14 However, these reports mainly rely on single or retrospective cases.
In NS and JMML, disease-causing mutations target various genes of the RAS/MAPK pathway.15 ,16 With heterozygous germline-activating missense mutations in up to 40% of cases, PTPN11 (MIM 176876) is the most commonly involved gene in NS.17 Most patients with NS and MPD harbour a PTPN11 mutation,14 ,18 and about 35% of sporadic JMML cases display an acquired somatic PTPN11 mutation.19
PTPN11 encodes the cytoplasmic phosphatase SHP2, which enhances the signal transduction of growth factors and cytokines by upregulating RAS/MAPK pathway activation. A functional classification of PTPN11 mutations has been proposed, based on their predicted consequence on SHP2 function.20–22 Most PTPN11 mutations found in NS and sporadic leukaemia affect residues located close to or within the N-SH2/PTP interaction domain and disrupt the autoinhibition of the catalytic domain, increasing the basal and/or stimulated phosphatase activity of SHP2.21 ,23
The department of Genetics of Robert-Debré hospital collects the majority of molecularly confirmed French NS patients, and is the reference laboratory for JMML in France. This privileged situation prompted us to investigate the incidence of MPDs in a large cohort of patients with NS. This study allowed us to estimate the overall incidence of haematological abnormalities in these children, describe their natural history, and discuss phenotype/genotype correlations.
Patients and methods
Patients
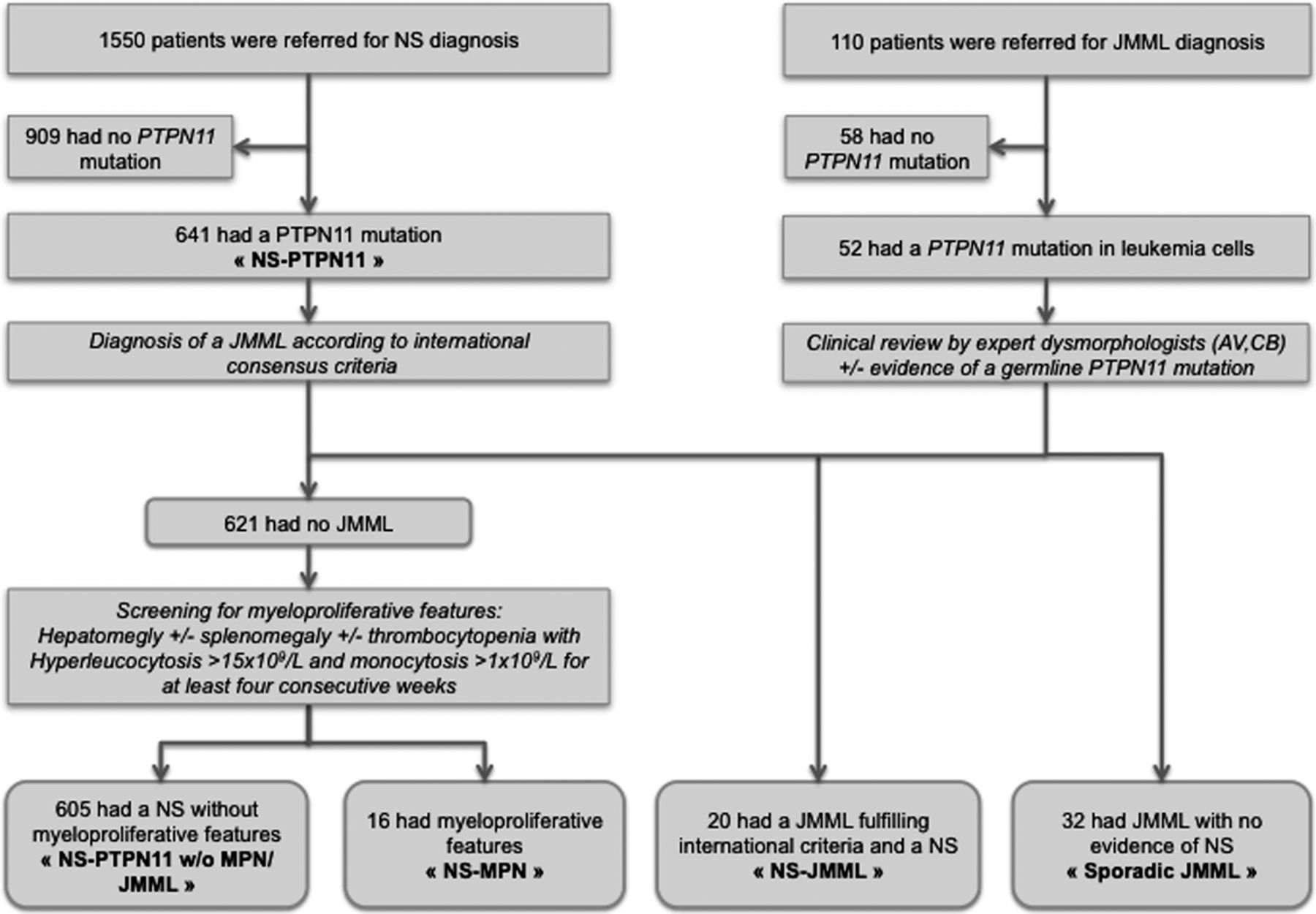
Patient samples were referred to the lab either for suspicion of NS or suspicion of JMML (figure 1). Written informed consent of the parents was obtained according to the declaration of Helsinki.

Flowchart and definition of the different groups of patients.
Between 2002 and 2012, we identified a PTPN11 mutation in leukaemic cells in 52/110 (47%) patients referred for JMML diagnosed according to international consensus guidelines.8 Among them, 20 had a diagnosis of NS based on published diagnostic criteria,24 reviewed by expert clinical geneticists (AV, CB). The clinical diagnosis was confirmed in 19/20 cases either directly by demonstrating a PTPN11 mutation in germline (non-haematopoietic) DNA, or indirectly by identifying a PTPN11 mutation in one of the parents (n=2). These patients were referred as the NS-JMML group. The remaining 32 patients with no clinical signs of NS and no constitutional PTPN11 mutation were designated as the sporadic JMML group (figure 1).
During the same period of time, we identified a germline PTPN11 mutation in 641 probands referred for molecular confirmation of NS, who represented the NS-PTPN11 group considered in this study. Clinical notes for NS patients (prenatal events, growth, malformations, developmental milestones…) were routinely collected on a printed form filled by the clinicians. The clinical form included, among others features, the presence of hepatomegaly or splenomegaly, and the presence of an abnormal blood count.
We focused on NS-PTPN11 patients presenting with splenomegaly, hepatomegaly and/or thrombocytopenia. Clinicians and biologists in charge of patients presenting with at least one of these signs were secondarily contacted for further details. Hyperleukocytosis >15×109/L with sustained monocytosis >1×109/L for at least four consecutive weeks, unrelated to infection, was confirmed in 16 patients. These patients showed myeloproliferation but did not fully meet the diagnostic criteria for JMML. They are hereafter referred to as NS-MPD patients (figure 1). DNA from non-haematopoietic tissue was obtained from 6/16 NS-MPD patients, allowing the confirmation of a germline PTPN11 mutation.
The diagnosis of JMML or MPD was confirmed by a cytomorphological review of blood and bone marrow smears by a single expert (OF) and by the demonstration of endogenous myeloid progenitor cell growth in all patients who were tested (27 sporadic-JMML, 14 NS-JMML and 7 NS-MPD).
Because of the small number of patients and the continuum between the two haematological phenotypes, NS-JMML and NS-MPD patients were grouped together as a NS-MPD/JMML group for statistical analyses of phenotype/genotype correlations.
Samples
Peripheral blood and/or bone marrow aspirates were collected on EDTA at diagnosis. For patients with JMML or MPD, non-haematopoietic tissues (fibroblasts, nails, amniotic fluid) were also collected. Genomic DNA was extracted using a Qiagen Mini or Midi Kit (Qiagen Gmbh, Hilden, Germany).
Sequence analysis
Mutation screening of PTPN11 was performed on genomic DNA by bi-directional Sanger sequencing of exons and their flanking intron-exon boundaries.19 ,25
SNP array analysis
SNP array analysis was performed using a GeneChip Human SNP Array 6.0 (Affymetrix, Santa Clara, California, USA) in paired leukocyte and fibroblast DNA samples. CEL files were created using Affymetrix GeneChip Command Console operating software and Genotyping Console 2.1, according to the manufacturer's protocols (Affymetrix). The Partek Genomics Suite was used for the analysis of both copy number alterations (CNA) and the loss of heterozygosity (LOH). Regions of LOH were detected using a Hidden Markov Model algorithm in the standard Partek workflow.
Exome sequencing
Targeted enrichment and massive parallel sequencing were performed on paired genomic DNA from leukocytes and fibroblasts. Exome capture was carried out using the SureSelect Human All Exon V4+UTRs (Agilent Technologies, Santa Clara, California, USA), and sequencing with a HiSeq2000 instrument (Illumina, San Diego, California, USA). Image analysis and base calling were performed using the Real Time Analysis (RTA) pipeline V. 1.14 (Illumina). The alignment of paired-end reads to the reference human genome (UCSC GRCh37/hg19) and variant calling were carried out using the CASAVA V.1.8 pipeline (Illumina). Variant annotation, SNP filtering (IntegraGen Exome databases, dbSNP135: (http://www.ncbi.nlm.nih.gov/projects/SNP), 1000 Genomes: (http://www.1000genomes.org) and HapMap: (http://hapmap.ncbi.nlm.nih.gov)) and patient-matched germline variant filtering were achieved using an in-house pipeline by IntegraGen (Evry, France).
Myeloid progenitor cell growth
In vitro growth assays of myeloid progenitors were performed by plating bone marrow and/or peripheral blood mononucleated cells in semisolid methylcellulose with and without leukocyte-conditioned medium (cytokine medium, LCM, StemCell Technologies, Vancouver, Canada).26 Colonies (aggregates containing >50 cells) were scored on days 11 and 14. ‘Endogenous growth’ corresponded to the presence of colony-forming units-granulo-monocyte (CFU-GM)>10 colonies or -monocyte (CFU-M)>5 colonies) in the absence of growth factors.
Clonality assay
Clonality was assessed in female patients by analysing X-chromosome inactivation patterns. The human androgen receptor gene (HUMARA) was assayed as previously described.27 Because a heterozygous pattern of methylation of the HUMARA locus has been reported in clonal haematopoietic cells, three additional loci (ZDHHC15, SLITRK4 and PCSK1N) were studied.28 ,29 Briefly, methyl-specific digestion of DNA was performed separately by 2 endonucleases (HhaI, HpaII). The four loci were subsequently amplified by PCR. Reaction products were run on an automated capillary sequencer (ABI 3130 Genetic Analyzer, ABI) using Genescan software (ABI). The ratio of inactive to active X-chromosome was derived from the allelic ratio in digested and undigested DNA.30 DNA samples from 60 healthy age-matched patients and 2 X-skewed patients were used as negative and positive controls, respectively. The assay was considered informative when at least 3 markers showed heterozygosity. X-inactivation was considered to be skewed when the ratio was >5 for at least one locus.
Statistical analysis
Statistical analyses were performed using R software V.2.14.0 (http://www.R-project.org, the R Foundation for Statistical Computing, Vienna, Austria) and GraphPad Prism V.5.01 for Windows (GraphPad Software, San Diego California USA). Comparisons of quantitative variables and distributions were made using a non-parametric Kruskall–Wallis test, and Fisher's Exact, and χ2 tests, respectively.
Results
Myeloproliferation in NS
We identified MPDs in 36/641 (5.6%) NS patients with a germline PTPN11 mutation. Twenty patients (3%) met the consensus diagnostic criteria for JMML (figure 1). Haematological anomalies most often appeared in the neonatal period, earlier than in patients with sporadic JMML (table 1). NS-MPD patients presented significantly milder clinical and biological anomalies than NS-JMML and sporadic JMML patients (table 1). Bone marrow aspiration of NS-JMML and sporadic-JMML patients showed similar cytological anomalies, that is, granular and/or erythroid hyperplasia. Monosomy 7 was not observed in any of the NS-JMML patients who were karyotyped (n=15).
Haematological features at diagnosis of NS-MPD, NS-JMML and sporadic PTPN11-JMML patients
Six patients with NS-JMML had dysgranulopoiesis, of which two had an excess of undifferentiated blast cells (see online supplementary figure 1). Bone marrow aspiration performed in three NS-MPD patients showed granular hyperplasia in two and lymphoid hyperplasia in one patient. Clonality analysis in girls with NS-MPD (n=6) or NS-JMML (n=5) demonstrated skewed X-inactivation suggestive of clonal haematopoiesis in 3/5 informative girls with NS-MPD and in 2/4 informative girls with NS-JMML (see online supplementary figure 2).
Clinical outcome
None of the NS-MPD patients required chemotherapy, and all are alive, with a median follow-up time of 3.0 years (table 2; figure 2). The median time between diagnosis and the last peripheral blood cell count was 38.8 months (min–max; 14.1–143.8). At this time point, monocytosis and/or thrombocytopenia persisted in 8/16 patients. Notably, patient #13 developed a hyperdiploid B-cell progenitor acute lymphoblastic leukaemia (BCP-ALL) at the age of two years.
Clinical presentation and follow-up of patients with NS-JMML and NS-MPD
{kind=link}
{kind=link}
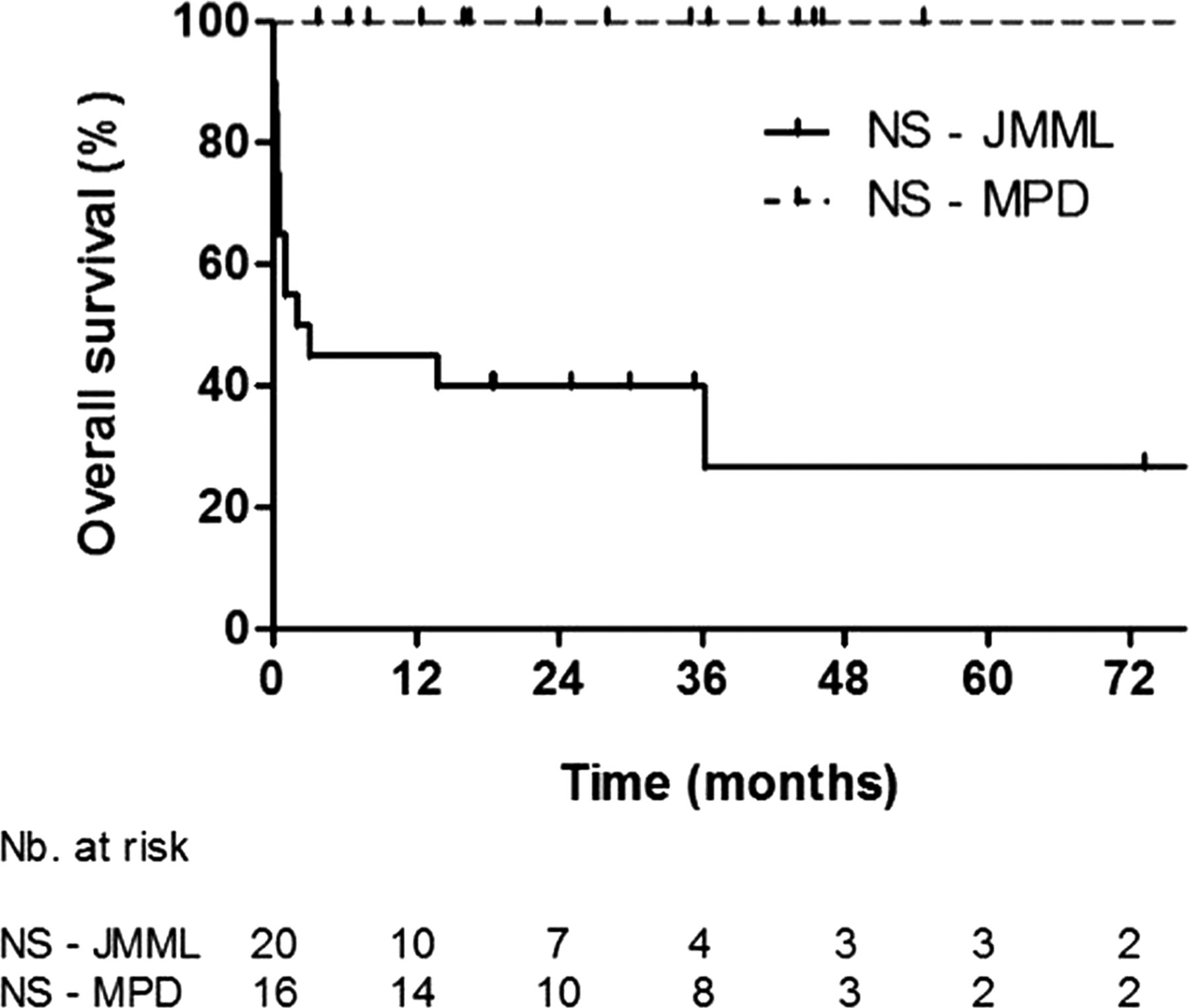
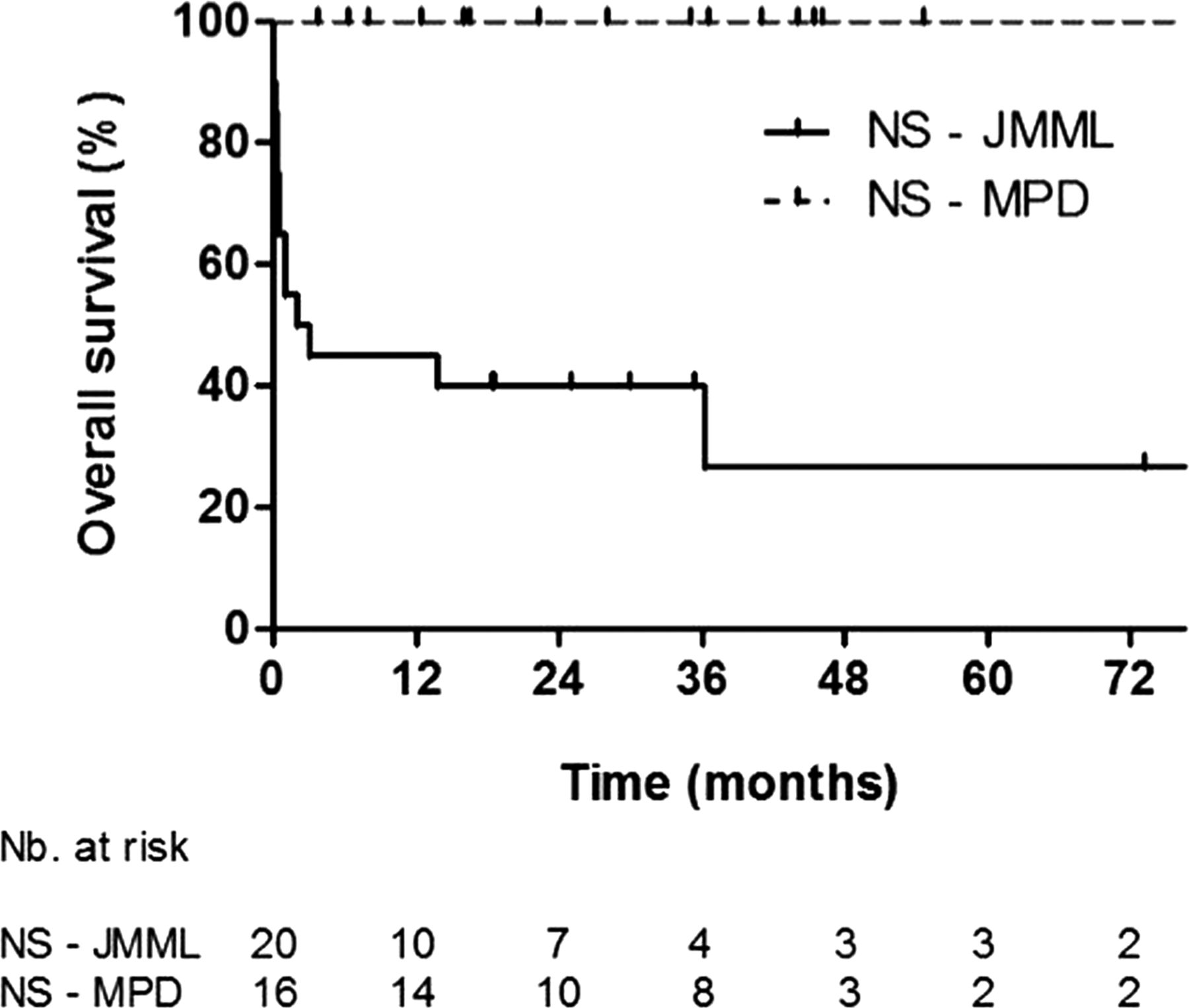
Kaplan–Meier curves of survival in patients with NS-MPD and NS-JMML.
Life-threatening complications related to congenital heart defects, pleural effusion, leukaemia infiltrates and/or thrombocytopenia were noted in 12/20 (60%) NS-JMML patients (table 2). Ten of these 12 patients died soon after diagnosis (median time ([min–max): 26.5 days (9–124)) from haemodynamic failure, respiratory failure or cerebral haemorrhage. Anti-tumoral chemotherapy (6-mercaptopurine, cytarabine and etoposide) was administered in two patients (#1 and #3). After a transient clinical improvement, both died in a context of rapid WBC rise or blast crisis. The other patients received only supportive care. Necropsies of patients #1, #8 and #10, who died of pulmonary failure, revealed major tumoral infiltration of the lungs, confirming the causal link between JMML and death. In patients #2, #3 and #5, pulmonary and cardiac failure were strongly suspected to have been increased by visceral tumoral infiltration, since the WBC count rose rapidly and reached its highest level on the day of death. For patient #4, a cerebral haemorrhage was diagnosed at 3 days of life in a context of thrombocytopenia (28×109/L) related to JMML. Patient #12 had an unusual evolution. He achieved complete remission (CR) of JMML according to standard criteria, but had persistent severe thrombocytopenia 1 year after CR. Repeated bone marrow smears confirmed amegakaryocytosis.
In the remaining eight patients with NS-JMML, haematological symptoms appeared between birth and 7 months, and were related to tumoral infiltrates in the viscera. None of these patients required intensive care at diagnosis or during follow-up. Three were treated with 6-mercaptopurine and reached CR (patients #14, #17 and #19). Patient #17 subsequently developed a neuroblastoma. Five patients who did not receive any chemotherapy had either a complete (#13 and #15) or partial (#16, #18 and #20) spontaneous remission. Three of them (#18, #19 and #20) died later, under circumstances not directly related to their initial haematological presentation, from either pulmonary infection, postallograft toxicity, or cardiac surgery complications.
With a median follow-up time of 2.8 years, the 2-year overall survival rate was, respectively, 40.0% (SE=11.0%), 100% and 61.8% (SE=9.6%) for NS-JMML, NS-MPD, and sporadic JMML (figure 2). Only 2/20 patients with NS-JMML received haematopoietic stem cell transplantation, versus 23/32 patients with sporadic JMML.
Interestingly, compared to the rest of the NS-PTPN11 cohort (n=605), NS-JMML and NS-MPD patients showed a higher rate of polyhydramnios (table 3). There was no difference based on gestational age or growth retardation; NS patients who developed MPD/JMML tended to have a more severe neonatal presentation with a higher frequency of hemodynamic and/or respiratory failure related to chylothorax and heart defects (table 3). Surprisingly, 11/12 (91.6%) patients with severe neonatal manifestations were males, which is significantly different from the sex-ratio in NS-PTPN11 patients (p=0.03)
Perinatal features of NS patients according to the presence of haematological manifestations
Pattern of PTPN11 mutations
We compared the pattern of PTPN11 mutations in 3 groups: (1) sporadic JMML patients; (2) NS-JMML/MPD patients and (3) NS-PTPN11 patients with no MPD (table 4). For 30/32 patients (93.7%), acquired somatic mutations were clustered in codons Asp61, Ala72 and Glu76. None of the substitutions identified in sporadic JMML was found in patients with NS. Mutations identified in NS-JMML/MPD patients overlapped with those of common NS patients, but some of them were over-represented. Among 5 NS patients with a p.Thr73Ile mutation, 3 had JMML and 1 had MPD. These 4 patients suffered a mild clinical course with the spontaneous improvement of haematological anomalies. The most frequent mutations, targeting codon Asp61, were also over-represented: 10/36 (28%) in NS-MPD/JMML versus 38/605 (6%) in NS patients (p<0.01). Among patients with these mutations, 10/48 {20.8%—5% CI 10.5 to 35.0%} developed MPD in early childhood (table 4). Strikingly, a p.Asp61His, reported once in AML,32 but never seen previously in NS, was found in two NS-JMML patients (#1, #3). These male patients suffered from a particularly aggressive form of neonatal JMML that rapidly led to death, after a blast crisis in one patient (table 2; online supplementary figure 1).
PTPN11 (NM_002834) mutations identified in sporadic JMML (n=32) and NS-JMML/MPD (n=36) in comparison with the prevalence of these mutations in NS patients who did not develop myeloproliferative disease (n=605)
Almost all PTPN11 mutations found in sporadic JMML cases affect the switch properties of the encoded enzyme, SHP2.21 Some mutations found in NS-JMML/MPD affect SHP2 catalysis or specificity (see online supplementary table 1). According to the classification proposed by Tartaglia and others,17 an excess of class I mutations is observed in NS-JMML/MPD patients, with a rate between that of sporadic JMML and NS.
Taken together, these data suggest that some germline PTPN11 mutations are more likely to induce myeloproliferation in infants. However, most PTPN11 mutations identified in NS-MPD/JMML patients were also found in NS patients with no haematological phenotype. For instance, only 2/133 (1.5%) patients with p.Asn308Asp had haematological anomalies. Moreover, in 4 patients (NS-JMML: #18, #2, #5 and NS-MPD: #5), the mutation was inherited from a parent with no history of MPD.
Pangenomic screening for a second hit
We thus investigated whether somatic genetic lesions could participate in the occurrence of JMML in NS patients. Twelve paired germline/leukaemia samples were analysed by SNP array and eight of these were further subjected to whole exome sequencing. We identified a subclonal duplication of the X-chromosome in one male patient. Another male patient with NS-JMML had a mosaic Klinefelter syndrome (patient #8: 47, XXY (13)/46, XY (7)). However, no compound heterozygosity or co-occurrence of a second germline mutation targeting genes known to be involved in RASopathies (NRAS, KRAS, HRAS, PTPN11, SOS1, BRAF, RAF1, CBL, SHOC2, SPRED1, NF1, RIT1) were identified in these eight NS-JMML cases.
Discussion
Children with NS are predisposed to a spectrum of haematologic abnormalities including MPDs, which may regress without treatment or follow a more aggressive clinical course resembling JMML.2 ,4 ,13 Little is known about the natural history of the disease and potential phenotype/genotype correlations. Several cases of NS-JMML have been reported individually, but the incidence of MPDs in NS has not been estimated. Kratz et al reported eight new NS-JMML/MPD patients and reviewed 11 cases from the literature, while Timeus et al33 reported three patients.14
We identified haematological features typically associated with myeloproliferation in 36/641 (5.6%) NS patients with a germline PTPN11 mutation. Twenty of them met the consensus diagnostic criteria for JMML and 16 had MPD. The strength of our study comes from the central position of our lab in the molecular diagnosis of both NS and JMML in France, which reduces the likelihood that only severe or haematological cases were examined. However, considering the weaknesses inherent in our a posteriori, declarative mode of ascertainment of MPD cases, and the absence of systematic haematological screening in NS patients, this incidence may be an underestimate.
The haematological features observed in our patients encompassed a broad phenotypic spectrum ranging from transient MPD to clinical courses similar to sporadic PTPN11-JMML. According to clinical and haematological data, patients were found clustered in two groups. The first group accounted for about 2% of NS cases and followed a mild clinical course. Some of these patients were transiently treated to control myeloproliferation but all underwent spontaneous remission. Most patients reported so far in the literature belong to this group, suggesting that NS patients do not develop severe JMML.14 However, our study identified a second group of patients who represented 3% of NS cases. These patients developed bona fide JMML, often with an excess of blasts and very severe neonatal manifestations. Half of them died in the first month of life. Few patients have been reported in the literature with similar features.21–23 We thus suspect that JMML may have been overlooked due to the early death of such infants in neonatal intensive care units, the presence of comorbidities and the lack of specific confirmatory tests. Intriguingly, the majority of patients with severe neonatal JMML were males.
NS-PTPN11 has a low infantile mortality even if some cases remain undiagnosed: fetuses terminated for incurable heart defects, or spontaneous fetal death due to hydrops. By contrast, the survival of NS-JMML patients is very poor (table 2). In our series of NS-PTPN11, JMML is the first cause of death in childhood. Whether cardiac or respiratory failure leading to death is a consequence of myeloproliferation and tumoral infiltrates in the lung, or whether mutations that induce myeloproliferation also induce a more severe general disorder remains an open but important question. The latter assumption may discourage aggressive leukaemia therapy in children whose prognosis is likely to remain poor.
It would thus be important to distinguish patients who will spontaneously recover from those who will have a more severe disease course. The endogenous growth of myeloid progenitors, a hallmark of JMML related to the selective hypersensitivity of these cells to GM-CSF, has been found in all patients who were tested, and cannot be used to predict the severity of haematopoietic complications. No difference in clonal patterns could be seen between patients with NS-JMML and NS-MPD, but the clonality assay suffers from limitations and must be interpreted with caution. An excess of blasts or blast crisis was shown in two male patients who experienced a severe clinical course, in line with previous observations.4 ,11 These observations strongly support the malignant nature of myeloproliferation in some patients.
The development of MPD and JMML in NS show striking similarities with other pretumoral syndromes in infants, such as transient acute myeloproliferation in Down syndrome, that can either spontaneously regress or evolve towards malignancy.34 However, while GATA-1 mutations mark myeloproliferation in Down syndrome, our exome and array studies did not identify any cooperative somatic mutation that could drive leukaemogenesis. Additionaly, no second germline genetic variation in NS genes was associated with the occurrence of JMML in NS patients, as previously described in patients with NS who underwent fetal death or early malignancy.35 ,36
PTPN11 mutations associated with NS-JMML/MPD were clustered in a limited number of amino acids. In line with previous reports,14 ,18 ,37 p.Thr73Ile mutation was strongly associated with haematological disorders. However, it was present in only 4/36 (11.1%) of NS-JMML/MPD patients of our cohort, whereas, it has previously been reported by Kratz et al14 to be the most common mutation in JMML/MPD (42%). However, these authors did not report patients with severe neonatal JMML leading to early death, suggesting that their patient ascertainment method was different and overlooked neonatal cases. This over-representation of the p.Thr73Ile mutation in previous studies could explain why patients with NS are often considered to develop mild MPD with spontaneous regression rather than bona fide JMML. Indeed, in our series, the clinical presentation varied from a mild course with spontaneous recovery for all p.Thr73Ile patients to rapidly fatal neonatal disease in patients with p.Asp61His. Mutant SHP2 proteins associated with malignancies are more activating than those observed in NS.20 ,21 This supports a model in which a higher gain-of-function threshold for SHP2 activity is required to induce leukaemia: the relatively mild gain-of-function effects of the SHP2 mutants associated with NS are usually inadequate to deregulate haematopoietic precursor cell proliferation, whereas, PTPN11 mutations found in isolated malignancies have higher gain-of-function effects incompatible with embryonic development. We confirmed this general observation in our patients: Asp61His, the only mutation observed in somatic leukemias and NS, caused a severe phenotype leading to the death of both patients soon after birth.
In line with murine models, our observations support the view of a strong endogenous role of a germline PTPN11 mutation in the occurrence of myeloproliferative complications,38 and the existence of high-risk mutations. Nevertheless, the risk of developing MPD or JMML cannot be precisely predicted by the underlying PTPN11 mutation, since only a fraction of patients with a given mutation will develop a haematological disease, even within a family.
In conclusion, NS may be associated with early onset, life-threatening haematological complications. RASopathies and MPDs are probably overlooked in cases of early lethality or in patients hospitalised in neonatal or paediatric intensive care units, and surely overlooked after a termination of pregnancy. Haematological evaluation should be performed systematically in newborns with severe haemodynamic or respiratory failure clinically suspected of having NS, without delaying these investigations until the presence of a mutation is confirmed. Our observations confirm that infants with NS-MPD should be managed conservatively unless refractory cytopenias, compromised vital organ functions, or an overt leukaemia with blast crisis develops.34 In particular, excessive hyperleucocytosis may be observed in certain clinical stressful situations, that is, during infections or after surgery, in NS-MPD patients, with no clinical symptoms and spontaneous decrease.
We recommend that all NS children be systematically evaluated for clinical signs of MPD (splenomegaly, hepatomegaly) and basic haematological parameters at least every 3 months during the first year of life, and twice during the second year of life. Of particular concern is the observation that two of the 22 NS children who survived MPD/JMML developed a second tumour later in childhood, suggesting that these children deserve a very close clinical follow-up.
Acknowledgments
SNP-array analyses were performed in the ‘Plateforme de Génomique constitutionnelle-Nord’ (PfGC-Nord) (J Soulier, S Quentin). We thank Jocelyne Vivent for help with clinical data collection. We also acknowledge the network of haematologists, neonatologists, geneticists, cytologists and pathologists who participated in the diagnosis and care of the children: Drs B Isidor, A David and S Denizot (Nantes), J Tandonnet, S Ducassou, M Micheau and N Aladjidi (Bordeaux), G Plat and S Julia (Toulouse), M Poirée and F Guiliano (Nice), F Cartault (St Denis-la Réunion), C Pondarre, MP Pages, and F Alsohim (Lyon), P Brosset (Limoges), E Kermorvant (Paris-Necker), L Jules and V Picard (Paris-Bicêtre), A Petit, A Coulomb and H Lapillonne (Paris-Trousseau), C Samson (Nouméa), B Leheup (Nancy), R Touraine (St Etienne), F Petit, M Holder-Espinasse and B Nelken (Lille), J Roume (St Germain-en-Laye) and B Doray, Patrick Lutz (Strasbourg).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
- Data supplement 2 - Online table
Footnotes
-
Contributors MS, AC, BC, SP and JL performed experiments and data analysis. SG and CA made the statistical analysis. OF performed cytomorphological review of blood and bone marrow smears. NP performed data management. CB, AC, NS, FM, IG, DA, CP, YB and AB were involved in local study implementation, patient recruitement and reviewed the manuscript. MS, AV, MZ, CC and HC wrote the manuscript. HC designed and supervised the study and the writing of the manuscript.
-
Competing interests None.
-
Ethics approval INSERM.
-
Provenance and peer review Not commissioned; externally peer reviewed.