Article Text

Abstract
Background Chronic mucocutaneous candidiasis disease (CMCD) may result from various inborn errors of interleukin (IL)-17-mediated immunity. Twelve of the 13 causal mutations described to date affect the coiled-coil domain (CCD) of STAT1. Several mutations, including R274W in particular, are recurrent, but the underlying mechanism is unclear.
Objective To investigate and describe nine patients with CMCD in Eastern and Central Europe, to assess the biochemical impact of STAT1 mutations, to determine cytokines in supernatants of Candida-exposed blood cells, to determine IL-17-producing T cell subsets and to determine STAT1 haplotypes in a family with the c.820C>T (R274W) mutation.
Results The novel c.537C>A (N179K) STAT1 mutation was gain-of-function (GOF) for γ-activated factor (GAF)-dependent cellular responses. In a Russian patient, the cause of CMCD was the newly identified c.854 A>G (Q285R) STAT1 mutation, which was also GOF for GAF-dependent responses. The c.1154C>T (T385M) mutation affecting the DNA-binding domain (DBD) resulted in a gain of STAT1 phosphorylation in a Ukrainian patient. Impaired Candida-induced IL-17A and IL-22 secretion by leucocytes and lower levels of intracellular IL-17 and IL-22 production by T cells were found in several patients. Haplotype studies indicated that the c.820C>T (R274W) mutation was recurrent due to a hotspot rather than a founder effect. Severe clinical phenotypes, including intracranial aneurysm, are presented.
Conclusions The c.537C>A and c.854A>G mutations affecting the CCD and the c.1154C>T mutation affecting the DBD of STAT1 are GOF. The c.820C>T mutation of STAT1 in patients with CMCD is recurrent due to a hotspot. Patients carrying GOF mutations of STAT1 may develop multiple intracranial aneurysms by hitherto unknown mechanisms.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
High susceptibility to mucosal and skin candidiasis is a hallmark of several primary immunodeficiency disorders (PIDs) associated with impaired interleukin (IL)-17 T cell immunity.1–7 Isolated chronic mucocutaneous candidiasis (CMC) was first described in the 1960s, but its genetic causes remained unknown until recently.4 ,6 CMC may occur in patients with PIDs and various other pathological conditions, such as severe combined immunodeficiency, combined immunodeficiency-like dedicator of cytokinesis 8 deficiency, signal transducer and activator of transcription (STAT) 3 deficiency causing autosomal-dominant hyper-IgE syndrome (AD-HIES), autosomal recessive (AR), autoimmune polyendocrine syndrome type 1, AR IL-12Rβ1 and IL-12p40 deficiencies, AR tyrosine kinase (TYK) 2 deficiency and AR caspase recruitment domain nine deficiency.1 ,6 However, CMC may be the only or the principal disease phenotype in patients with PIDs, such as AD IL-17F and AR IL-17RA deficiencies,8 and gain-of-function (GOF) mutations of STAT1.9 In such cases of essentially isolated CMC, the patients are described as suffering from CMC disease (CMCD).8–13
Mutations affecting the STAT1 coiled-coil domain (CCD) were the first to be implicated in CMCD, initially in a patient with sporadic CMCD from Ukraine and then in various kindreds of different ethnic origins.9–13 This was somewhat surprising, because previously reported STAT1 mutations affecting the DNA-binding domain (DBD), the SH2 domain, the tail segment and even some other mutations affecting the CCD (K201N and K211R) had been linked to Mendelian susceptibility to mycobacterial disease (MSMD), with or without predisposition to viral, but not candidal diseases.7 It was suggested that various CCD mutations, including the D165G mutation found in the index patient from the Ukraine and the 11 additional sequence variants found in CMCD patients, actually resulted in a gain of phosphorylation and GOF due to a loss of dephosphorylation of STAT1 homodimers after translocation to the nucleus.9 The picture has been complicated even further by recent reports of two patients with sporadic CMCD and the T385M DBD mutation associated with a GOF due to hyperphosphorylation and impaired dephosphorylation after nuclear translocation.13 Indeed, previously reported mutations affecting the DBD were loss-of-function (LOF) and were found in patients with mycobacterial disease.7
Various STAT1 GOF mutations (affecting the CCD or the DBD) have been systematically associated with susceptibility to CMCD. Primary STAT1 defects have been described over the last 10 years, and the susceptibility of affected patients to infections with viruses, mycobacteria, staphylococci and fungi has been shown to depend on the monoallelic or biallelic and GOF or LOF nature of the mutations they carry.7 Several patients with GOF STAT1 mutations presenting not only CMCD, but also severe viral infections and/or autoimmune signs have recently been described.9–13 Clearly, more clinical data and phenotype–genotype studies are required to define the CMCD caused by STAT1 sequence variants. We describe here the genetic, immunological and clinical findings for nine patients with STAT1 mutations and CMCD diagnosed in four Eastern and Central European (ECE) countries by whole-exome sequencing (WES) and Sanger sequencing.
Patients and methods
Patients
The patients and their families are described in the online supplementary case reports. All the patients are Caucasian. Genetic data, infectious signs and immunological findings are summarised in tables 1⇓–3. All the studies described here were approved by the Regional Ethics Committee of the University of Debrecen Medical and Health Science Center and by the institutional review boards of the centres at which the patients were managed. Informed consent was obtained from the patients or their parents (for minors).
Genetic features of patients with STAT1 mutation and chronic mucocutaneous candidiasis diagnosed in Eastern and Central Europe
Infectious manifestations in new patients reported in this study with STAT1 mutations and chronic mucocutaneous candidiasis*
Immunoglobulins, antibodies and white blood cells
Detection of mutations
Exome sequencing. DNA (3 μg) was extracted from Epstein–Barr virus (EBV)-transformed B cells (EBV-B cells; see below) and sheared with a Covaris S2 Ultrasonicator. An adaptor-ligated library was prepared with the TruSeq DNA Sample Preparation Kit (Illumina). Exome capture was performed with the SureSelect Human All Exon kit (Agilent).9 ,14–17 Paired-end sequencing was carried out on an Illumina HiSeq 2000 generating 100 bp reads. For sequence alignment, variant calling and annotation, the sequences were aligned with the human genome reference sequence (GRCh37 build), with the Burrows–Wheeler Aligner.17 ,18 Downstream processing was carried out with the Genome analysis toolkit (GATK) and SAMtools.18 ,19 Variant calls were made with a GATK Unified Genotyper. All calls with a read coverage two times or less and a Phred-scaled quality of 20 or less were excluded. All variants were annotated with an in-house annotation software system.
Sanger sequencing. Genomic DNA from the patients was prepared with the Gen Elute Blood Genomic DNA kit (Sigma-Aldrich). Targeted sequencing was performed by amplifying the exons and flanking intron regions of STAT1 by PCR. The PCR primers and sequencing primers used are available on request. Amplicons were sequenced with the Big Dye Terminator cycle sequencing kit (Applied Biosystems, Foster City, California, USA) and were analysed with an ABI 3130 capillary sequencer (Applied Biosystems, Foster City, California, USA). Sequence variants were identified relative to a reference sequence, GenBank accession no. ENST00000361099 for the STAT1 cDNA, in which the c.1 position corresponds to the A of the ATG translation initiation codon. Mutations are designated as recommended by den Dunnen and Antonarakis.20
Luciferase reporter assay
U3C cells, a STAT1-deficient fibrosarcoma cell line,9 were dispensed into 96-well plates (1×104 cells/well) and transfected with 100 ng/well reporter plasmids (Cignal GAS Reporter Assay kit, SA Biosciences) and plasmids carrying the c.537C>A or c.854A>G alleles of STAT1 or a mock vector, in the presence of Lipofectamine LTX (Invitrogen).9 Six hours after transfection, the cells were transferred back to medium containing 10% fetal bovine serum (FBS) and cultured for 18 h. The transfectants were then stimulated by incubation with interferon (IFN)-γ (10 or 1000 U/ml) for 16 h and subjected to luciferase assays with the Dual-Glo luciferase assay system (Promega). Experiments were performed in triplicate. The data are expressed as fold induction with respect to unstimulated cells.
Generation of EBV-transformed lymphoblasts
Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation through Ficoll-Paque PLUS (GE Healthcare Bio-Science AB, Uppsala, Sweden). They were then washed twice with phosphate buffer solution (PBS) and twice with Hanks balanced salt solution (Sigma). Cells (107/ml) were resuspended in 2.5 ml RPMI supplemented with 10% FBS, 1% glutamine and 1 µg/ml cyclosporin A (CyA). They were then infected with 2.5 ml of the supernatant of B95–8 cells infected with EBV and incubated for 2 h in a water bath at 37°C. We added 5 ml RPMI-1640 supplemented with 10% FBS, 1% glutamine and 1 µg/ml CyA, transferred the cells to T25 flasks (TPP, Switzerland) and incubated them at 37°C under an atmosphere containing 5% CO2 for 3 weeks.
Extraction of nuclear proteins and western blotting
EBV-B cells were stimulated with IFN-γ, IFN-α or IL-27 for 30 min and subjected to nuclear extraction with the Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Scientific) according to the manufacturer's instructions. We assessed phosphorylation by stimulating EBV-B cells with 2000 U/ml IFN-γ.9 In selected experiments, cells were stimulated with IFN-α (2000 U/ml) or IL-27 (2000 U/ml). The cells were then washed and incubated with 1 μM staurosporine in RPMI for 15 and 30 min, and nuclear proteins were extracted with the Protein Fractionation Kit (Thermo Scientific). The proteins were subjected to immunoblot analysis. The effect of the phosphatase inhibitor pervanadate on IFN-γ-induced STAT1 phosphorylation was assessed by incubating the transformed B cells with this compound for 5 min before stimulation as previously described.9
Candida
Candida albicans (ATCC 10231) was maintained on Sabouraud dextrose agar at 4°C, with subculture once monthly, by transfer onto fresh medium and overnight culture at 37°C.21 Stationary-phase cultures were prepared by inoculating 2 ml of Dulbecco's modified Eagle's Medium (DMEM) supplemented with 100 U/ml penicillin and 10 µg/ml streptomycin with Candida. The density of Candida was adjusted to 1×104 cells/ml with a McFarland densitometer. Heat-inactivated yeast was prepared by inoculating 5 ml SalSol (TEVA, Debrecen, Hungary) with Candida and incubating the culture at 56°C for 1 h. We checked that the heat inactivation was effective by transferring various dilutions of Candida suspension onto Sabouraud dextrose agar and incubating for 48 h.22 The heat-killed Candida suspension was then centrifuged at 4000×g for 10 min, and the cell pellet was resuspended in DMEM. The density of heat-inactivated Candida was adjusted to 1×106/ml with a McFarland densitometer.
Cell isolation and stimulation
Peripheral blood samples were obtained from the patients and controls. PBMCs were incubated as described above. After centrifugation and several washes in Krebs-Ringer phosphate buffer, the cells were resuspended in DMEM (Sigma-Aldrich, St Louis, Missouri, USA). PBMCs were incubated (5×105 cells/well) for 48 h at 37°C in 96-well round-bottomed culture plates that had been sterilised by gamma irradiation and were free of pyrogens, RNA, RNases, DNA and DNases (TPP, Trasadingen, Switzerland), either alone or in the presence of heat-killed C albicans. Once the blood cells had been exposed to Candida for 2 days, the plates were centrifuged and the supernatants were removed and stored at −20°C for the determination of cytokine concentrations.
Enzyme-linked immunosorbent assays
The concentrations of IFN-γ, IL-1β, IL-6, IL-17A and IL-22 in the supernatants of Candida-exposed PBMCs and IL-17F in the supernatants of cultured lymphocytes were determined by sandwich enzyme-linked immunosorbent assays (Quantikine, R&D Systems, Minnesota, USA). All experiments were performed in duplicate or triplicate.
Induction and differentiation of IL-17-producing T cells
PBMCs were washed with SalSol (TEVA) and suspended in RPMI-1640 supplemented with 10% FBS and 1% glutamine. Adherent cells were removed by incubation for 4 h in RPMI, in T25 flasks (TPP), at 37°C, under an atmosphere containing 5% CO2. The non-adherent cells were activated with anti-CD3 antibodies in a 12-well plates and were treated with a cocktail of human transforming growth factor-β1 (5 ng/ml), IL-1β (10 ng/ml), IL-6 (50 ng/ml) and recombinant human (rh)IL-23 (20 ng/ml), all from PeproTech (London, UK). Two days later, an additional 2 ml of RPMI containing the cytokines listed above, plus IL-2 (22 ng/ml; PeproTech), was added to each well. Four days later, another 2 ml of RPMI without cytokines was added to the wells. The following day, the cells were treated with brefeldin A isolated from Penicillium brefeldianum (1 μg/ml; Sigma) and were left unstimulated or were stimulated with phorbol 12-myristate 13-acetate (PMA; 50 ng/ml) and ionomycin (IMC; 10−5 M) for 6 h at 37°C under an atmosphere containing 5% CO2, centrifuged at 150 ×g for 6 min, suspended and washed in 2% FBS–bovine serum albumin (BSA)–PBS buffer.
Flow cytometry
We detected intracellular IL-17 and IL-22 in CD3 and CD4 T cells with an Accuri C6 flow cytometer (Becton Dickinson). CD3 cells were identified with an fluorescein isothiocyanate (FITC)-conjugated mouse anti-hCD3 IgG1 mAb (Beckman Coulter), and CD4 cells were labelled by incubation with allophycocyanin-conjugated mouse anti-hCD4 IgG1 mAb (BD) for 30 min at 4°C. The cells were then incubated with 50 μl Fix&Perm Fixation Medium A (Invitrogen) for 15 min at 4°C. They were then washed with 2% FBS–BSA–PBS buffer and resuspended in 50 μl Fix&Perm Permeabilization Medium B (Invitrogen). Intracellular IL-17 was detected with phycoerythrin-conjugated mouse anti-hIL-17 IgG1 mAb, and intracellular IL-22 was labelled with FITC-conjugated mouse anti-hIL-22 IgG1 mAb, both from R&D Systems. Cells were incubated for 30 min at 4°C, washed with 2% FBS–BSA–PBS and suspended in 500 μl 0.1% paraformaldehyde.
Haplotype analysis
Familial haplotyping was performed by Sanger sequencing as described above. The following previously reported intronic polymorphisms were genotyped: rs7597768, rs2280234, rs2280233, rs2066804, rs41507345 and rs11693463. The primers used for PCR and sequencing are available upon request.
Results
Genetic findings
We carried out WES, to identify sequence variation, in patients P8 and P9 (table 1). The mutations detected were confirmed by Sanger sequencing. In patients P1–P7, Sanger sequencing was used to analyse STAT1. Two familial and five sporadic STAT1 mutations were found; all but three of these mutations were located in exon 10 of STAT1 and all were heterozygous, missense mutations (table 1). Some of the data for patients P4, P5 and P9 have been reported before,9 ,11 whereas the data for patients P1–P3 and P6–P8 are reported for the first time here. Two new STAT1 mutations affecting the CCD of STAT1 (patients P1 and P7) and one mutation affecting the DBD (patient P8) were detected. We isolated genomic DNA from the EBV-transformed lymphoblasts of patients P4, P5 and P8 and used it for Sanger sequencing. The mutations found in freshly isolated blood cells were also present in EBV-transformed cells (see online supplementary figure S1).
The two new c.537C>A (N179K) and c.854A>G (Q285R) mutations of STAT1 result in a GOF for GAF-dependent cellular responses
The two new mutations affecting the STAT1 CCD, N179K and Q285R, were found in a Czech patient (patient P1) and a Russian patient, respectively (P7). These patients were not available for biochemical studies. We therefore analysed the possible pathological consequences of these mutations by site-directed mutagenesis and studies of γ-activated factor (GAF)-dependent cellular responses to IFN-γ. We transfected STAT1-deficient U3C cells with wild-type (WT) STAT1 and c.537C>A (N179K) and c.854A>G (Q285R) STAT1 mutant constructs, and with the previously characterised CMCD-causing c. 821G>A (R274Q)9 and autosomal-dominant MSMD-causing Y701C STAT1 alleles Hirato O et al (manuscript submitted). Responses to cytokine stimulation were investigated by measuring the luciferase activity of the reporter gene under the control of the γ-activated sequence (GAS) promoter. Stimulation with various concentrations (10 and 1000 U/ml) of IFN-γ resulted in responses that were two to three times stronger in cells transfected with the two new mutant alleles or with the CMCD-causing R274Q allele than in those transfected with WT or MSMD-causing Y701C alleles (figure 1). These data suggest that the new heterozygous alleles reported in this study are GOF for GAF-dependent cellular responses to IFN-γ, one of the key STAT1-activating cytokines.

Induction of GAS-dependent reporter gene transcription activity in U3C cells stimulated with IFN-γ. The response to IFN-γ stimulation was measured by determining the activity of a luciferase reporter gene under the control of the GAS promoter. GAS transcriptional activity is shown as fold induction after 16 h of stimulation with IFN-γ (10 U/ml, left columns; 1000 U/ml, right columns), with respect to unstimulated cells. Mock and MSMD-causing Y701C allele-transfected cells served as negative controls. Upon stimulation with IFN-γ, cells transfected with either the N179K or the Q285R allele responded two to three times more strongly than those transfected with the WT or Y701C alleles. Similar results were obtained when cells were transfected with the GOF R274Q allele known to cause CMCD. CMCD, chronic mucocutaneous candidiasis disease; GAS, γ-activated sequences; GOF, gain of function; IFN, interferon; MSMD, Mendelian susceptibility to mycobacterial disease; WT, wild-type.
The T385M and R274W STAT1 mutations result in a gain of phosphorylation of STAT1
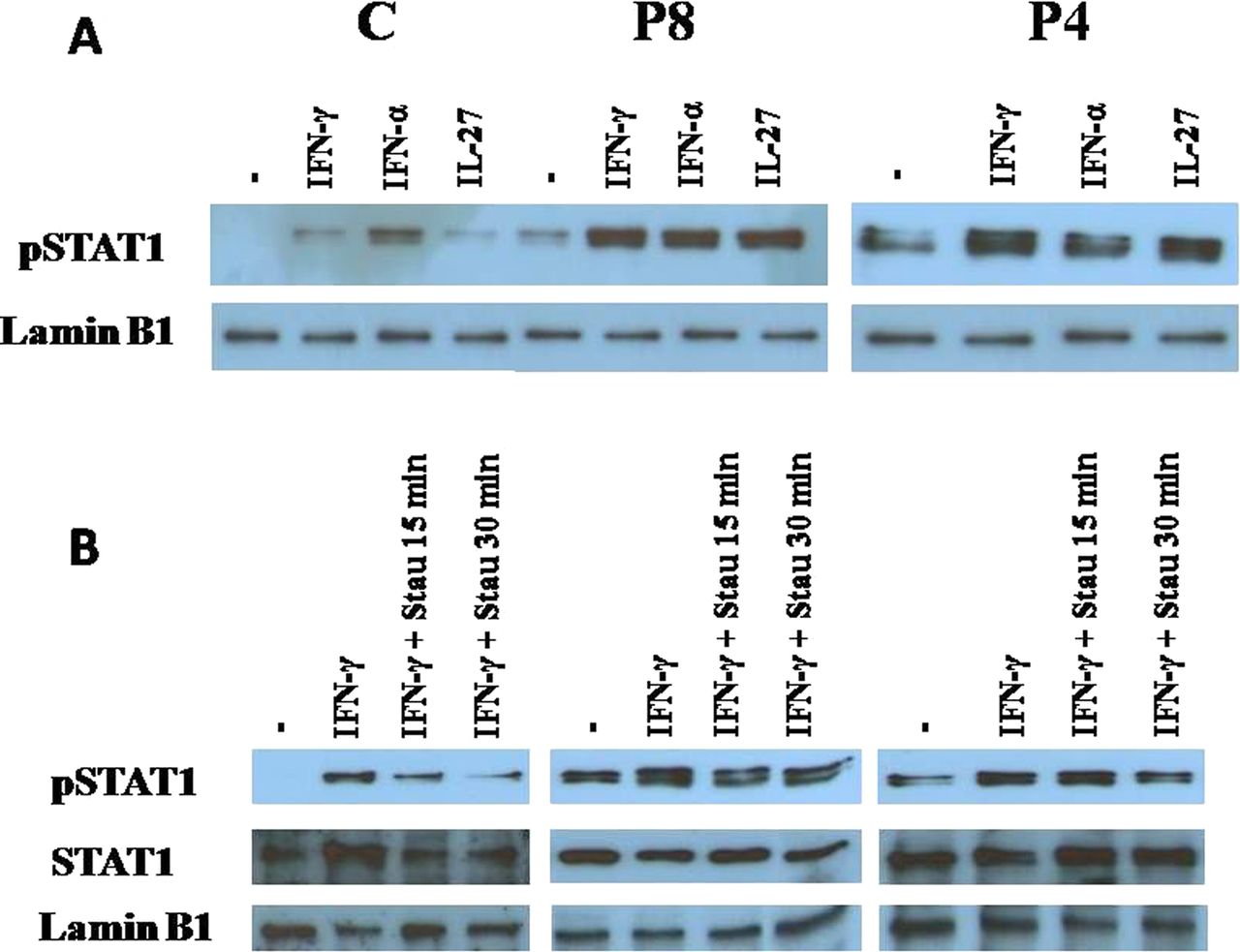
It was initially suggested that the STAT1 mutations in CMCD patients affected the STAT1 CCD only, resulting in a GOF because of a gain of phosphorylation.9 We detected a recently reported mutation, c.1154C>T (T385M), affecting the STAT1 DBD, in one of the Ukrainian CMCD patients (table 1; patient P8).13 EBV-B cells from P4 and P8, heterozygous for the R274W and T385M mutations, respectively, were treated with IFN-γ, IFN-α and IL-27, all of which predominantly activate STAT1.9 Western blotting showed that treatment with these cytokines resulted in stronger STAT1 phosphorylation in the nuclear extracts of cells from both patients than in those from controls (figure 2A), demonstrating that the heterozygous alleles were associated with a dominant phenotype of gain of STAT1 phosphorylation. Incubation with the TYK inhibitor staurosporine and comparison with the control lymphoblasts showed that the dephosphorylation of IFN-γ-treated EBV-B cells heterozygous for the STAT1 T385M or R274W alleles was impaired (figure 2B). By contrast, after pervanadate treatment, the phosphorylation of T385M STAT1 and R274W STAT1 was similar to that of WT STAT1 (see online supplementary figure S2). These data confirm that the T385M and R274W amino acid substitutions in STAT1 result in a gain of STAT1 phosphorylation due to a loss of dephosphorylation.9 ,13

STAT1 dephosphorylation in nuclear extracts. Western blotting was performed on nuclear extracts of EBV-transformed lymphocytes from P8 and P4, carrying the c.1154C>T (T385) and c.820C>T (R274W) heterozygous missense mutations of STAT1, respectively (table 1). (A) Before nuclear extracts were prepared, cells were treated with 2000 U/ml IFN-γ, 2000 U/ml IFN-α or 50 μg/ml IL-27 for 30 min. The data shown are representative of three independent experiments. The T385M (P8) and R274W (P4 and P5) STAT1 proteins are more strongly phosphorlylated than the WT protein in control cells. (B) Effect of the tyrosine kinase inhibitor staurosporine on STAT1 dephosphorylation in nuclear extracts of cells from healthy controls (left), P8 (middle) and P4 (right) with CMCD. EBV-transformed lymphocytes were treated with 2000 U/ml IFN-γ for 30 min and then with staurosporine (concentration, 8 ng/ml) for 15 and 30 min. Staurosporine induced the dephosphorylation of nuclear STAT1 in control cells but not in cells from P8 and P4. C, control; EBV, Epstein–Barr Virus; IFN, interferon; P, patient; STAT, signal transducer and activator of transcription; WB, western blot.
Release of cytokines by Candida-exposed white blood cells from CMCD patients
We measured the release of various inflammatory cytokines by freshly isolated PBMCs after 48 h of stimulation with heat-killed Candida. Similar amounts of IFN-γ, IL-1β and IL-6 were released by the cells of P8 (T385M) and control cells (figure 3A). By contrast, PBMCs from P8 released negligible amounts of IL-17A and IL-22, much smaller than those released by Candida-exposed cells from healthy controls (figure 3B). Similar data were obtained with cells from P4 and P5 carrying the R274W allele (figure 3C,D). These data suggest a normal ‘Th1-type’ reaction, and normal IL-1β and IL-6 release in response to stimulation with C albicans, but an impaired ‘Th-17-type’ response to this fungus in patients with CMCD-associated mutations.

Secretion of cytokines by blood mononuclear cells. (A) IFN-γ, IL-1β and IL-6 secretion by mononuclear blood cells (5×105) from P8 (see table 1) and unrelated healthy controls, as measured by enzyme-linked immunosorbent assays (ELISA), after stimulation with heat-killed C albicans (105) for 48 h. (B) Secretion of IL-17A and IL-22 by blood mononuclear cells (5×105) from P8 and unrelated healthy controls, as measured by ELISA, after stimulation with heat-killed Candida (105) for 48 h. (C) Cells from the patient and controls secreted similar amounts of IFN-γ, whereas (D) the patient's cells secreted much smaller amounts of both IL-17A and IL-22 than control cells. The data shown are representative of two experiments performed in triplicate. C, control; NS, non-stimulated; P, patient.
Ex vivo differentiation of IL-17-producing and IL-22-producing T cells
PBMCs were isolated and treated with IL-17-inducing cytokines as described above. Flow cytometry was used to determine the percentages of CD3 T cells and CD4 T cells-producing cytoplasmic IL-17 or IL-22 (figure 4). Before marker analysis, the cells were treated with PMA and IMC for 6 h. Consistent with the cytokine-release data, the patient with the T385M STAT1 allele displayed impaired development of IL-17-producing and IL-22-producing T cells (figure 4A–C). Similar results were obtained for the cells of one of the patients (P4) with the heterozygous R274W allele (figure 4A–C).

Ex vivo development of IL-17-producing and IL-22-producing T cells in P8 with the c.1154C>T STAT1 mutation. Percentages of CD3/IL-17+ (A), CD4/IL-17+ (B) and CD4/IL-22+ (C) cells, as determined by flow cytometry after incubation for 6 h with phorbol 12-myristate 13-acetate and ionomycin. At least three independent experiments were carried out in each case. C, control; P, patient.
In another study, we evaluated the in vitro differentiation of IL-17-producing T cells from naïve CD45RA CD4 T cells under various conditions of stimulation (plates coated with anti-CD3 antibody, soluble anti-CD28 antibody alone or together with various combinations of IL-1β, IL-23 and IL-6) (Okada S. et al, manuscript submitted). We found that the best conditions were as follows: plates coated with anti-CD3 antibody, soluble anti-CD28 antibody plus IL-1β and IL-23. Under these conditions, we observed strong impairment of the development of IL-17-producing T cells in CMCD patients bearing STAT1 GOF mutations, as assessed by comparison with controls.
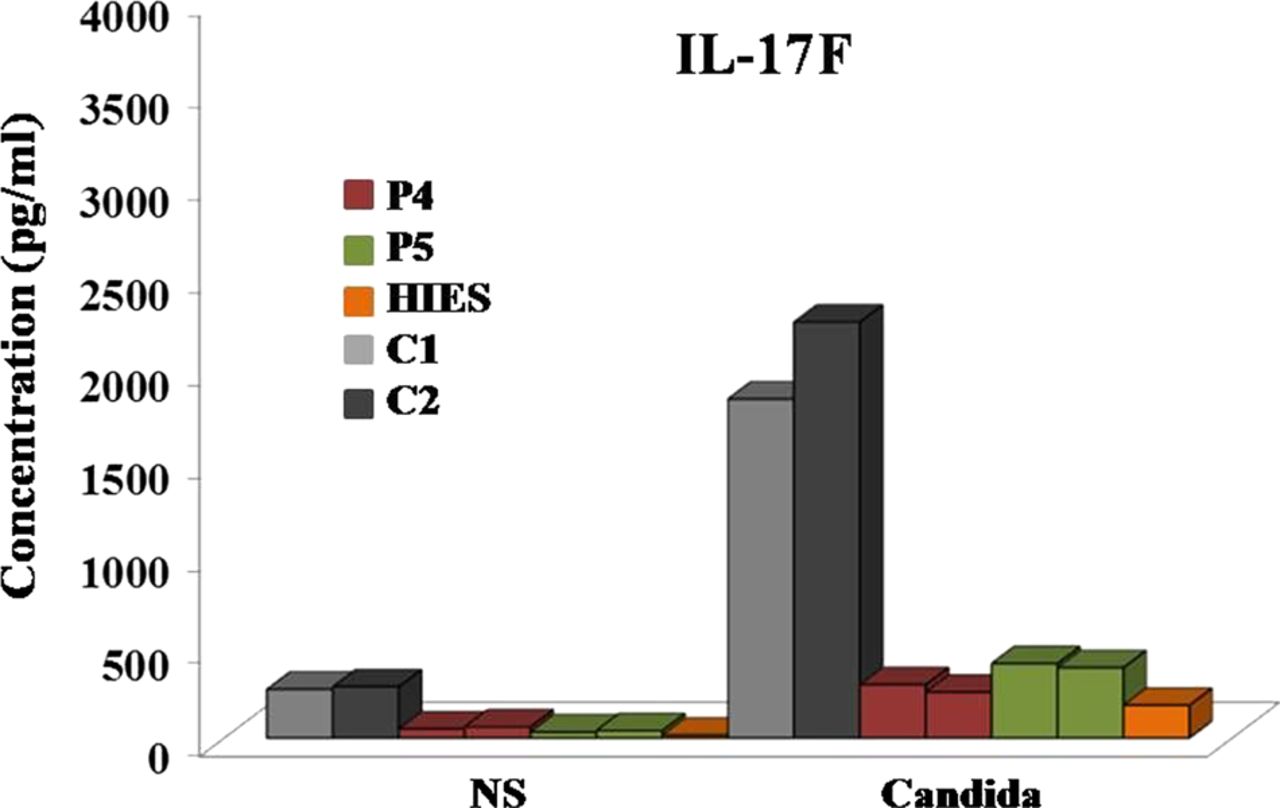
We also measured the release of IL-17F by PBMCs from P4 and P5 after the cytokine-induced in vitro differentiation of T cells into IL-17-producing lymphocytes. After 5 days of cytokine treatment, the cells were stimulated with PMA and IMC for 6 h and IL-17F concentrations were determined by enzyme-linked immunosorbent assays (ELISA) on the supernatants. Control experiments were performed with cells isolated from healthy individuals and with cells obtained from a patient with AD-HIES caused by a dominant negative mutation of STAT3 (figure 5). An impairment of IL-17F release by the cells of both CMCD patients bearing the R274W allele and by the cells of the patients with AD-HIES was observed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Concentrations of IL-17F in the supernatant of lymphocytes after 5 days of differentiation in vitro. Cells were treated with transforming growth factor-β1, IL-1β, IL-6, rhIL-21, rhIL-23 and IL-2, as described in ‘Patients and methods’ section. IL-17F levels were determined, by enzyme-linked immunosorbent assays, in the supernatant of cells from two healthy controls, patients P4 and P5 with the R274W allele, and a patient with hyper-IgE syndrome caused by a heterozygous dominant negative c.994C>T (H332Y) mutation of STAT3. C, control; NS, non-stimulated; P, patient.
Hotspot mutation affecting the STAT1 CCD
The sequencing of STAT1 in two related patients revealed the presence of a c.820C>T heterozygous mutation affecting the CCD and leading to a predicted R274W substitution in the STAT1 protein (table 1). About 30% of the CMCD patients with STAT1 mutations have mutations affecting R2749–11 13 (Okada S et al, manuscript submitted). It was not clear whether the mutations at this position had arisen de novo in all families, or whether the large number of families carrying this mutation resulted from a founder effect. We therefore investigated whether the two mutations affecting position 274 (c.820C>T, leading to the R274W substitution, and c.821G>A, leading to the R274Q substitution) were due to a founder effect or a hotspot, by genotyping six polymorphisms surrounding the mutation in healthy and affected members of seven families (table 4). We obtained evidence of a hotspot effect for both mutations in families in which the mutation was absent from both healthy parents and occurred de novo in the patient. The affected individuals from different families had different haplotypes, ruling out the founder effect proposed by another group.10
Haplotype analysis of STAT1 mutant allele
Complications of CMCD
Autoimmune thyroiditis appears to be the most common autoimmune complication in CMCD patients9 12 13 (supplementary case reports). No signs of systemic autoimmune disease were observed. Five patients reported here developed oesophageal candidiasis, and P2, P4 and P8 also had oesophageal stricture, necessitating balloon dilatation (P4) or Nissen fundoplication (P8). Recurrent pneumonia developed in six patients and was complicated with pulmonary abscess (P4), bronchiectasis and even clubbing (P9).1 Four patients had recurrent mucocutaneous herpes simplex virus (HSV) infection and two patients with the R274W allele (P4 and P5) also had recurrent shingles.11 These findings suggest that Candida oesophagitis, damage to the respiratory tissues and recurrent herpes virus disease may be common complications of CMCD and that early antifungal prophylaxis might help to prevent these complications. P1 suffered a fatal complication in the form of mycotic cerebral aneurysm leading to intracranial bleeding.
Discussion
The pathogenesis of CMC in several complex PIDs, such as AD-HIES with LOF STAT3 mutations,23 24 and that of AD-CMCD with STAT1 GOF mutations, has only recently been determined.9–13 ,25 This report provides the first comprehensive overview of the molecular genetics, immunological and clinical features of patients diagnosed with sporadic and familial CMCD in ECE. We describe here several new STAT1 alleles with mutations affecting the CCD, and new phenotypic manifestations of the T385M mutation affecting the DBD. The two new mutations reported here (N179K and Q285R) increase the number of known CMCD-causing STAT1 mutations to 15. All but one of the heterozygous CMCD-causing mutations, including the two new sequence variants described in this cohort, affected the CCD of STAT1, consistent with previous suggestions that this is the most frequently affected domain of the protein in patients with CMCD (table 1).9 ,10 The two patients with previously unknown CCD mutations were not available for biochemical testing. However, site-directed mutagenesis and GAS-dependent reporter gene expression assays clearly suggested that the underlying mechanism involved a GOF for GAF responses to IFN-γ. The higher level of reporter gene induction presented here indicates that these new mutations may be responsible for the CMCD phenotype, consistent with previously published data showing CMCD-causing GOF mutant STAT1 alleles to be dominant for GAF activation.9 P8, like two recently described Japanese patients,13 had mutations affecting the DBD of STAT1. We found that the mutation in P8, like the new CCD mutations, resulted in the impaired dephosphorylation of STAT1. The artificial F172W mutation, positioned next to the N179K mutation found in patient P1, and the artificial G384A or G384W mutations next to the T385M mutation, described elsewhere13 and found in patient P8 have been reported to impair the dephosphorylation of STAT1.26 ,27 However, loss of dephosphorylation may not be the only mechanism responsible for the GOF, and other mechanisms, such as stronger binding of the mutant DBD STAT1 to GAS, may also play a role.
We suggest that determinations of the Candida-stimulated release of IL-17A and IL-22 cytokines by PBMCs is a reliable and reproducible method for assessing the impairment of IL-17 T cell-mediated immunity in CMCD patients (11 and this report). In our hands, live and heat-killed C albicans were equally effective for inducing cytokine release by control blood mononuclear cells (only the data for heat-killed Candida are shown). The data presented here suggest that the IL-17 T cell deficiency responsible for the greater susceptibility to mucosal Candida infection in CMCD patients is not limited to the cells beneath body surfaces, instead extending to circulating blood cells. In contrast to the impaired release of IL-17A and IL-22 by PBMCs from patients with STAT1 GOF mutations affecting the CCD or DBD, the Candida-induced release of IFN-γ, IL-1β and IL-6 was similar in the blood cells of patients and controls, suggesting that the activation of alternative cytokine pathways was intact. Intriguingly, it has been reported that PBMCs exposed to Candida-derived mitogens produce smaller amounts of IFN-γ in patients with STAT1 GOF alleles10 ,12 By contrast, we showed that the production of IFN-γ by patients’ PBMCs stimulated with heat-killed Candida blastoconidia was similar to that of control subjects. These discrepancies may reflect differences in the assay systems used. We believe that our data are more consistent with clinical observations, as IFN-γ does not play a non-redundant role in immunity to C albicans, whether systemic or peripheral. Indeed, patients with inborn errors of IFN-γ do not present candidiasis of any form.28 Consistent with the impaired release of IL-17A, IL-17F and IL-22, ex vivo analysis by using flow cytometry showed IL-17 T cell levels to be lower in CMCD patients with mutations affecting the CCD or the DBD than in controls. Thus, mutations affecting either the CCD (patient P4) or the DBD (patient P8) of STAT1 result in similar immunological phenotypes and cytokine release profiles in terms of IL-17 T cell immunity.
The phenotypic manifestations of the various STAT1 mutations affecting the CCD may differ, despite the similar molecular and cellular/immunological features. P1 developed multiple intracranial aneurysms and intracranial bleeding. Two of the 47 patients described in the first report on CMCD and STAT1 GOF mutation also developed cerebral aneurysms at the ages of 8 and 38 years.9 One of the four patients described in a subsequent report died from cerebral vasculitis.13 However, no cerebral vascular complications were reported in subsequently described patients,10–12 ,25 suggesting that CMCD patients may develop this complication only rarely. It has been suggested that intracranial aneurysms due to the damage caused by Candida result directly from candidal invasion of the arterial wall, usually in major intracranial vessels.29–31 Recent findings suggest that STAT1 production and function may be enhanced in the tissues of abdominal aortic aneurysms, and that tyrosine-phosphorylated STAT1 levels may be higher in these tissues, resulting in higher levels of total STAT1 protein activity.32 It remains unclear whether GOF STAT1 mutations play a direct role in aneurysm formation in CMCD patients or whether aneurysm formation is a direct consequence of the damage caused by Candida.33 ,34 P1 had not only multiple intracerebral aneurysms, but also renal abscesses caused by C albicans, suggesting that patients with STAT1 mutations affecting the CCD may develop deep-seated candidal disease. This is the first such observation to be reported for patients with CMCD. The clinical phenotype of P8 was somewhat different from that previously described for two Japanese patients carrying the same mutation (T385M).13 P8 had early oesophageal involvement and developed oesophageal stricture necessitating Nissen fundoplication. He also presented recurrent severe HSV infections. None of these clinical features were observed in the Japanese patients, who were otherwise of similar age and genotype. By contrast, both Japanese patients had recurrent lower respiratory tract infections, bronchiectasis and autoimmune thyroiditis. One of these patients died from disseminated intravascular coagulation and pulmonary insufficiency of unknown origin. These data suggest that the phenotypic expression of the T385M allele may differ significantly between affected patients.
The enhanced response to type I IFNs in the STAT1 GOF patients is reminiscent of the type I signature observed in patients with autoimmune diseases, such as Aicardi–Goutières syndrome patients or patients with immuno-osseous dysplasia spondyloenchondrodysplasia.35 ,36 These patients also present autoimmune thyroiditis. It is therefore possible that the enhanced response to type I IFNs is responsible for the autoimmune thyroiditis observed in the patients with STAT1 GOF mutations. In addition, patients treated with recombinant type I IFNs often present such autoimmune manifestations as secondary effects.37 ,38 Furthermore, higher levels of intracellular adhesion molecule-1 (ICAM-1) expression have been implicated in the pathogenic mechanism of autoimmune thyroiditis.39 ICAM-1 levels have never been assessed in patients with STAT1 GOF, but in vitro studies of FRTL-5 thyroid cells exposed to IFN-γ showed an induction of ICAM1 gene transcription via GAS binding to the ICAM1 promoter.40 ,41 It is therefore possible that STAT1 GOF mutations predispose patients to autoimmune thyroid disease by enhancing the responses of thyroid cells to type I IFNs.
Autoimmune hepatitis, which may also occur in patients with STAT1 GOF mutation, is known to be mediated by T cells, and CD4 T cells are the predominant population of lymphocytes infiltrating the liver.42 It has been shown that, in a mouse model, IFN-γ activation of STAT1 contributes to Con-A-induced liver damage by activating CD4 cells and inducing hepatocyte death.43 Based on these observations, we suggest that STAT1 GOF mutation may cause autoimmune hepatitis by inducing a strong IL-12β-IFN-γ- Janus kinase–STAT1 circuit in affected patients.
The R274W substitution found in two members of the Hungarian family resulted from one of the most frequent STAT1 mutations, suggesting that R274 may be a hotspot for mutation in patients with CMC.9 ,11 However, a previous report on STAT1 sequence variants suggested that affected patients may have a founder mutation.10 We therefore tested the hypothesis of a founder effect by carrying out haplotype analyses on the two patients described here and six previously described families.9 Haplotype analyses of two families carrying the R274W mutation suggested that this mutation was associated with a common disease-causing allele, but we demonstrated that R274 was a hotspot for mutation in patients with CMCD (table 4). Not only did we observe the de novo occurrence of the mutation in two families, but we also provide genetic evidence that the mutation is carried by different alleles in patients from different families. The de novo occurrence of mutations affecting other positions in the CCD of STAT1 (Y170N and D165H) has already been described.9 Overall, these data, together with the high incidence of mutations affecting this domain of STAT1, provide strong evidence that this DNA region is prone to errors. The increasing number of CMCD cases reported in publications over the last year might be considered suggestive of a founder effect, but we demonstrate here that this DNA sequence may be prone to germline replication errors. However, the reasons for this DNA sequence being a ‘super’ hotspot for mutation despite its lack of enrichment in trinucleotide repeats remain to be elucidated.
In summary, we report here molecular and clinical data for the nine patients with CMCD diagnosed to date in ECE countries. WES and Sanger sequencing revealed two previously unknown alleles of STAT1 with mutations affecting the CCD domain. Immunological and biochemical studies have shown that both CCD and DBD mutations may result in impairment of the Candida-stimulated release of IL-17A/F and IL-22 by lymphocytes, and that CMCD patients have impaired IL-17 T cell differentiation ex vivo. We also report a young patient with a previously unknown STAT1 mutation and multiple intracranial aneurysms and intracranial bleeding, suggesting that inflammatory vascular disease may be a phenotypic consequence of STAT1 GOF alleles affecting the CCD. Our data suggest that the R274W substitution is due to a mutation hotspot rather than a founder mutation. Further studies are required to define the precise molecular mechanisms of the complications observed in CMCD, including, in particular, the role of Candida and Candida-derived molecules in the development of aneurysms. Furthermore, the contribution of memory T cell defects to the recurrent HSV disease found in four patients in this cohort remains unclear, and the possible relationship between autoimmune thyroiditis and central or peripheral defects of controlling autoimmunity remains to be defined. Specific approaches, such as the J Project, are required to investigate PID gene mutations and genotype–phenotype relationships in various populations.44–47 (Horváth Z. et al manuscript in press).
Acknowledgments
We thank Alexandra Bársony for technical assistance, and Yelena Nemirovskaya and Zsuzsa Horváth for assistance in the preparation of the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online CaseReports
- Data supplement 2 - Online figures
Footnotes
-
Contributors BS performed the flow cytometry, western blot and genetic experiments. BT, GC, SO, SC, AP, J-LC, LM and AA performed and evaluated molecular genetic studies. ST and AKS performed ELISA assays. NS, AB, PR, DN, LC, ZT, AS, JL and LM took care of patients and designed immunological and clinical diagnoses. LM designed the study and wrote the paper.
-
Funding This work was supported in part by the UD Faculty of Medicine Research Fund (Bridging Fund 2012) and the TÁMOP 4.2.2.A-11/1/KONV-2012–0023 ‘VÉD-ELEM’ project to LM, St Giles Foundation grant to JLC, and the National Center for Research Resources and the National Center for Advancing Sciences (NCATS) grant number 8UL1TR000043 from the National Institutes of Health to JLC and SC, and ANR grant number GENCMCD 11-BSV3–005-01 to AP. SC was supported by the AXA Research Fund.
-
Competing interests None.
-
Ethics approval Approved by the Regional Ethics Committee of the University of Debrecen Medical and Health Science Center and by the institutional review boards of the centres at which the patients were managed.
-
Provenance and peer review Not commissioned; externally peer reviewed.