Article Text

Abstract
Background Graves’ disease is a female preponderant autoimmune illness and the contribution of the X chromosome to its risk has long been appreciated. However, no X-linked susceptibility loci have been indentified from recent genome-wide association studies (GWAS).
Methods We re-examined the X chromosome data from our recent GWAS for Graves’ disease by including males that were previously excluded from the X chromosome analyses. The data were analysed using logistic regression analysis including sex as a covariate, and an additive method assuming X chromosome inactivation, implemented in snpMatrix.
Results A cluster of single nucleotide polymorphism (SNPs) at Xq21.1 was found showing association with genome-wide significance, among which rs3827440 was a non-synonymous SNP of GPR174 (Plogistic regression= 9.52×10−8; PsnpMatrix=4.60×10−9; OR=1.76, 95% CI 1.45 to 2.13). The association was reproduced in an independent sample collection set including 4564 Graves’ disease cases and 3968 sex matched controls (combined Plogistic regression=5.53×10−21; combined PsnpMatrix=4.26×10−22; OR=1.69, 95% CI 1.53 to 1.86). Notably, GPR174 was widely expressed in immune related tissues and rs3827440 genotypes were associated with distinct mRNA levels (p=0.002). GPR174 did not show sex biased gene expression in our expression analysis. Resequencing study suggested the contribution of some rare variants in the GPR174 gene region to disease risk with a collapsing p value of 1.16×10−3.
Conclusions The finding of an X-linked risk locus for Graves’ disease expands our understanding of the role of the X chromosome in disease susceptibility.
- Genetics
- Genome-wide
- Complex traits
- Thyroid disease
- Endocrinology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Consistent with many other autoimmune illnesses, Graves’ disease exhibits a pronounced gender bias, with a female to male ratio of about 5:1 in the Chinese population, similar to that in Caucasian populations.1 ,2 The X chromosome is partly responsible for the hyperresponsiveness of the female immune system. It is reasonable to presume that some genes located on the X chromosome may play an important role in the susceptibility of Graves’ disease. However, recent genome-wide association studies (GWAS) did not reveal any loci on the X chromosome to be associated with this disorder.3 ,4 Reviewing the published GWAS for complex diseases showed that the focus of GWAS and subsequent meta-analyses has been on the autosomes, whereas X chromosomal data have usually been collected but not analysed.5–8 One reason for the neglect of the X chromosome in GWAS results could be the lack of a consensus analytical approach taking into account the specific features of X chromosomal data.
The special features of the X chromosome make tests for association less straightforward than those for autosomal chromosomes in mixed sex population studies.6–9 A female has two X chromosomes, while a male has only one. It should be noted that the pseudo-autosomal region (PAR) of the X chromosome has an homologous region on the Y chromosome, where loci are inherited like autosomal loci. Additionally, one X chromosome in females undergoes the X chromosome inactivation (XCI) process to maintain equal expression between sexes.10 The traditional approach is to stratify by sex, and several ways have been proposed for combining evidence across strata.9 Recently, Clayton proposed an additive test in which modelling was performed in the context that one of the female X chromosomes is inactivated.5 However, all these methods did not appear to have gained widespread use in GWAS analysis.8
In our previous study, 1536 Graves’ disease cases and 1516 sex matched controls of the Chinese Han population were genotyped using Illumina Human 660-Quad BeadChips including 14 141 chromosome X single nucleotide polymorphisms (SNPs).4 In total, 10 925 X chromosome SNPs in 1468 cases and 1490 controls matched criteria for quality control. The X chromosome data were investigated using the Cochran–Armitage test for trend ignoring males entirely, but no significant association at the X chromosome was found. In the current work, we expand our recent GWAS of Graves’ disease to include males when studying the X chromosome and follow-up our results using a larger independent case–control sample.
Methods
Samples and clinical characteristics
As described in our previously published data,4 1536 Graves’ disease cases and 1516 sex matched controls were recruited for stage 1 of the GWAS. In the current work, an additional 4564 Graves’ disease cases and 3968 sex matched controls were recruited for the replication study. A subset of samples from Shandong Province including 2608 cases and 2328 sex matched controls were sequenced for the GPR174 gene region. All individuals were of Chinese Han descent and provided informed consent with protocols approved by the local institutional review board. Diagnosis of Graves’ disease was based on documented clinical and biochemical evidence of hyperthyroidism, diffuse goitre, and the presence of at least one of the following: positive thyroid stimulating hormone (TSH) receptor antibody tests, diffusely increased I-131 (iodine-131) uptake in the thyroid gland, or exophthalmos. All individuals classified as having Graves’ disease were interviewed and examined by experienced clinicians.
Genotyping and quality control
DNA samples from 1536 Graves’ disease cases and 1516 controls were genotyped using Illumina (San Diego, California, USA) Human660-Quad BeadChips at the Chinese National Human Genome Center in Shanghai, China.4 After quality filtering of samples as described previously,4 1468 Graves’ disease cases and 1490 controls were used in the current analysis. The estimated genomic inflation factor was 1.02, indicating that overall population structure had negligible impact on the case–control association results. Therefore, we did not correct for population stratification in the association analysis. While examining the data of 14 141 X chromosome SNPs assayed in this study, we first eliminated the results of ‘heterozygous’ genotypes in males likely due to genotyping errors. Sequentially, we discarded five markers with Hardy–Weinberg equilibrium p value <10−6 in female controls, 870 with high missing call rates (>2%), and 2341 with minor allele frequency <1%, leaving 10 925 SNPs for subsequent analysis. The quality control procedure was performed with PLINK.11
Replication samples were genotyped for rs3827440 with TaqMan SNP Genotyping Assays (C_25954273_10) using the ABI 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, California, USA). The data were analysed using the ABI Prisms SDS V.2.1 software package.
Statistical analysis
After quality control, we used the genotypes of 10 925 SNPs in 1468 cases and 1490 controls for association analyses using two methods. The logistic regression analyses were performed in PLINK entering sex as a covariate.11 To perform a joint analysis for rs3827440 across the GWAS and replication stages, we used logistic regression analysis adjusted for gender and study stages to compute the p value. The one degree of freedom test described by Clayton was performed in snpMatrix.5 Under this test, the hemizygous males were treated as equivalent to the corresponding homozygous females at non-PAR X chromosome loci. The heterozygous females were modelled as half of the risk as the hemizygous males or homozygous females carrying the risk genotype. The p values calculated by snpMatrix for two stages were combined using Fisher's test. In females, the ORs of SNPs were estimated as the ORs of the homozygotes. In males, the ORs of SNPs were estimated as the per allele ORs. In the mixed sex samples, the hemizygous males were treated as equivalent to the homozygous females and the ORs were estimated from the comparison between the combined homozygous females and corresponding hemizygous males. A Mantel–Haenszel common OR was calculated across the two sample sets.
After excluding SNPs which did not pass the quality control filters, we imputed untyped and/or missing SNPs separately in cases and controls using the software IMPUTE212 and 1000 Genomes Phase I integrated variant set (March 2012) as reference. SNPTEST v2 was used to test for association with disease for genotyped and imputed SNPs (probability >0.9) under a logistic regression model with sex as a covariate. The effects of rare variants were assessed using collapsing methods.13 Because the observed variants were rare and, consequently, the females homozygous for the rare allele were extremely rare, the homozygous and heterozygous females and the hemizygous males were collapsed together, and a 2×2 table was constructed. We tested the difference between the proportions of individuals with rare variants in cases and controls using Fisher's exact test.13 All statistical analyses were performed with R (V.2.13) and SPSS software (V.17.0 for Windows) unless specified.
Tissue/cell gene expression patterns
We examined the expression of GPR174 in 16 different human tissues. cDNA samples of 15 tissues were from the Human Immune System Multiple Tissue cDNA (MTC) Panel and Human MTC Panel I (Clontech, Palo Alto, California, USA). In addition, Human Thyroid Total RNA (Clontech) was reverse transcribed using reverse transcription PCR (RT-PCR) reagents with random hexamers (Promega, Fitchburg, Wisconsin, USA) in accordance with the instructions of the manufacturer. The control cDNA contained in Clontech human MTC Panels were used as the positive control. Quantitative RT-PCR was performed using SYBR Green (TaKaRa, Otsu, Japan) in each 20 μl reaction containing 2 μl cDNA template on an ABI PRISM 7900 Sequence Detector (Applied Biosystems) with SDS V.2.1 software. GAPDH was used as an endogenous control. Primer sequences used are shown in online supplementary table S3. PCR products were visualised on a 3% agarose gel to confirm correct band sizes (see online supplementary figure S1). Each reaction was performed in duplicate, with final calculations resulting from means of duplicate wells. Normalisation for cDNA quantity was performed with GAPDH for each template and final abundance figures were adjusted to yield an arbitrary value of one for levels of gene specific expression in leucocytes using the ΔΔcq method.14
Quantification of allelic variation in gene expression
A total of 185 individuals including 141 females and 44 males were recruited for gene expression analysis. We drew 3 ml of peripheral blood from individuals participating in the study under fasting conditions. Genomic DNA was isolated from whole blood by the FlexiGene DNA Kit (Qiagen, Hilden, Germany). The genotypes of rs3827440 were determined by directed sequencing using Applied Biosystems 3730 platform. Sixty-four females with rs3827440 CC genotype and 43 females with TT, as well as 20 males with rs3827440 C allele and 19 males with T allele, were included in allele specific expression analysis. The females heterozygous for rs3827440 were excluded from allele specific analysis to avoid the influence of skewed XCI.
The RNA extraction was carried out using the QIAamp RNA Blood Mini Kit (Qiagen). Total RNA were reverse transcribed using RT-PCR reagents with random hexamers (Promega, Fitchburg, Wisconsin, USA) in accordance with instruction of the manufacturer. Quantitative RT-PCR was performed using SYBR Green (TaKaRa, Otsu, Japan) on an ABI PRISM 7900 Sequence Detector (Applied Biosystems) with SDS V.2.1 software. Each reaction was performed in triplicate, with final calculations based on the means of triplicate wells. GAPDH was used as an endogenous control. Primer sequences used are shown in online supplementary table S3. The ΔΔCq method was used to determine the expression levels of GPR174 for each sample.14 Mean threshold cycle (Cq) was calculated for each sample from three replicates and then used to calculate relative expression level (ΔCq), which is the difference between GPR174 Cq and GAPDH Cq. A median ΔCq value in the samples was used as a calibrator and the ΔΔCq was calculated using ΔCq of each sample minus the calibrator. The relative quantity of each sample was calculated using the relative quantification (RQ) formula (RQ=2−ΔΔCq). Distribution of relative gene expression levels was compared among males and females with different genotypes of rs3827440 by unpaired two-tailed Student t tests, respectively. In the combined samples, the difference of expression levels with genotypes was tested using an analysis of variance model adjusted for gender.
Resequencing
The GPR174 gene region was resequenced using the Applied Biosystems 3730 platform (see online supplementary table S4 for primers). We analysed traces using Phred, Phrap and Consed15 ,16 and identified variants with Polyphred.17 The variants identified were confirmed by sequencing the amplicons in both forward and reverse directions.
Results
A cluster of SNPs in strong linkage disequilibrium (LD) showed significant association in our GWAS samples when analysed using either of the two methods (figure 1A and online supplementary table S1). The associated SNPs were located near or within the G protein-coupled receptor 174 (GPR174) gene on Xq21.1 (figure 1B). The most significant association signal was observed at rs5912838 (Plogistic regression=4.60×10−8; PsnpMatrix=1.36×10−9; OR=1.80, 95% CI 1.48 to 2.18; see online supplementary table S1), which lies about 155 kb distal to GPR174. No significant signals were observed in the previously reported two Graves’ disease linkage regions, namely Xq21.33-q2218 ,19 and Xp1120 ,21; GPR174 locates midway between these two regions. Association analysis of imputed genotype data did not provide superior additional associated SNP in this region (see online supplementary table S2). Among the GWAS hits, a non-synonymous SNP rs3827440 (r2=0.93 to rs5912838) within GPR174 was an obvious functional variant of interest, though not presenting the top p value (Plogistic regression=9.52×10−8; PsnpMatrix=4.60×10−9; OR=1.76, 95% CI 1.45 to 2.13; figure 1B and table 1). Subsequently, rs3827440 was genotyped in an additional sample of 4564 Graves’ disease cases and 3968 sex matched controls. The association of rs3827440 to Graves’ disease was confirmed in the replication collection set and reached genome-wide significance in the combined analyses (combined Plogistic regression=5.53×10−21; combined PsnpMatrix=4.26×10−22; OR=1.69, 95% CI 1.53 to 1.86; table 1). The OR of rs3827440 across the two sample sets is 1.69 (95% CI 1.53 to 1.86), which is only lower than that of the SNP rs4947296 in the HLA gene region (OR=1.77, 95% CI 1.65 to 1.91),4 establishing GPR174 as an important locus with regard to the genetic susceptibility to Graves’ disease in the Chinese population.
Association results for rs3827440 using two methods

X chromosome-wide association results and regional plot of association results at Xq21.1. (A) X chromosome-wide association results calculated by using two methods. Values of −log10 p are plotted against chromosome positions. Purple and green dots represent p values calculated using Clayton's method by snpMatrix and logistic regression analysis by PLINK, respectively. (B) Association results of single nucleotide polymorphisms (SNPs) in genome-wide association study samples at Xq21.1. p Values were calculated using Clayton's methods. The colour of each genotyped SNP spot reflects its r2 to rs3827440 (large red diamond), changing from red to white. Genetic recombination rates, estimated by using the HapMap CHB (Han Chinese in Beijing, China) and JPT (Japanese in Tokyo, Japan) samples, are shown in cyan. Physical positions are based on NCBI (National Center for Biotechnology Information) build 37 of the human genome.
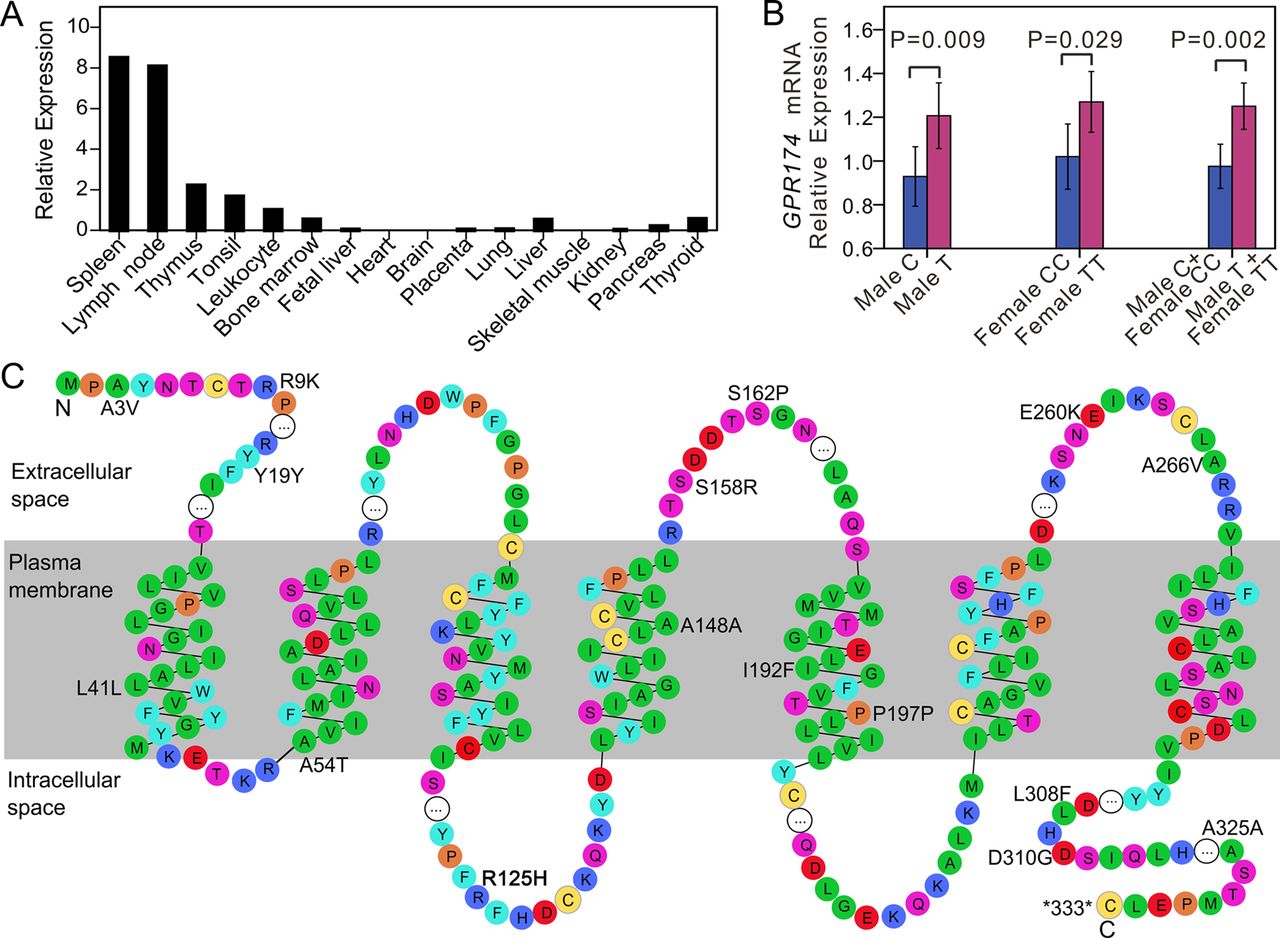
Since the expression profiles of most G protein coupled receptors (GPCRs) are unique, a highly selective tissue expression pattern may provide a clue with respect to receptor function.22 Therefore, we investigated the expression level of GPR174 in multiple human tissues/cells. GPR174 is widely expressed and has especially high expression levels in immune related organs and cells, including spleen, lymph nodes, thymus, tonsil, leucocytes, and bone marrow (figure 2A). Of note, although over 90% of GPCRs are expressed in the brain,22 no expression of GPR174 was observed in this organ. Moderate expression of GPR174, however, was detected in the thyroid tissue.
{kind=link}
{kind=link}
Expression analysis of GPR174 and distribution of the coding variants in GPR174. (A) Expression profiles of GPR174 in various human tissues by real-time reverse transcriptase PCR (RT-PCR). We performed real-time quantitative RT-PCR reactions in duplicate and plotted the means. Normalisation for cDNA quantity was performed by comparison with GAPDH controls and plotted as arbitrary relative expression units, where the leucocytes’ RNA expression level is equal to 1. (B) Relative expression levels of GPR174 against the distinct genotypes of rs3827440 were measured in peripheral blood cells (PBCs) from 39 males (C, n=20; T, n=19) and 107 females (CC, n=64; TT, n=43). Error bars,±SD. (C) Domain structure of GPR174 protein and the distribution of the coding variants. The structures are based on UniProtKB entry Q9BXC1 and the figure was prepared using RbDe. Amino acid residues are coloured according to residue types (red: acidic; blue: basic; purple: neutral hydrophilic; green: aliphatic; cyan: aromatic; orange: imino; yellow: thiol containing). The white circles represent contiguous stretches of amino acid residues which are omitted from this diagram.
SNP rs3827440 is a nucleotide transition (519C>T) in the single exon of GPR174 that causes the amino acid substitution P162S. Two SNPs, rs3810711 and rs3810712, both being located in the 5′ untranslated region (UTR) of GPR174, were in perfect LD with rs3827440 in our re-sequencing data. Real-time RT-PCR analysis revealed a significant correlation between expression levels of GPR174 in freshly isolated peripheral blood cells (PBCs) and the genotypes of rs3827440. Both female homozygous carriers and male carriers of the risk allele T were associated with a higher level of GPR174 expression (p=0.009 and p=0.029, respectively). When the combined homozygous and hemizygous T allele carriers were compared with the combined C allele carriers, the difference in expression levels gave a p value of 0.002 (figure 2B). These results suggest that rs3827440 and/or one or more variants in strong LD with rs382440 (eg, rs3810711 and rs3810712 in the 5′UTR) could influence GPR174 expression, thereby leading to the association with Graves’ disease. Of note, the P162S substitution in GPR174 maps to the second extracellular loop region (figure 2C), which is required for ligand recognition and receptor activation,23 and may therefore alter these activities.
Since genes with sex biased expression are enriched on the sex chromosomes,24 we also investigated whether GPR174 has sex biased expression. Although the mRNA level of GPR174 was slightly higher in the PBCs of females compared to those of males, no significant differences were observed (p=0.54 for TT females vs T males; p=0.09 for CC females vs C males; p=0.08 for combined females vs combined males, respectively).
Recent resequencing studies on complex diseases revealed common alleles of modest effect and rarer alleles with more considerable impact coexisting in the same disease genes.25 To investigate whether there are rare variants in GPR174 associated with risk to Graves’ disease, we sequenced the exon region and the 5′ as well as 3′ UTRs of GPR174 in 2608 Graves’ disease cases and 2328 controls. We identified 22 novel variants in addition to the three common ones (rs3810711 and rs3810712 in 5′ UTR, and rs3827440, table 2 and figure 2C). All of the 22 variants were very rare (minor allele frequency <0.5%), and none of them was previously listed in the dbSNP database. Of the 16 coding variants, 10 were non-synonymous variants. Although nine rare variant carriers were found among the 1272 female controls, no rare variants were observed in 1,056 male controls. Using a collapsing method,13 we tested the difference between the proportions of individuals with rare variants in cases and controls. The result suggested an enrichment of rare variants in cases with a p value of 1.16×10−3, but the evidence is not robust.
Rare variants in GPR174 indentified in 2608 cases and 2328 controls from resequencing
Discussion
The X chromosome spans about 155 million base pairs and contains more than 1000 genes; however, the X chromosome data have received surprisingly little attention in the wave of GWAS.5 ,7 ,8 Although several models for the X chromosome association analysis have been proposed and assessed, neither the traditional stratification analysis nor the newly developed methods appear to have gained widespread use in GWAS analysis.7–9 Moreover, the calculation of the ORs for X chromosome SNPs in sex mixed case–control studies was often missed in the literature.5 ,9 It is worth noting that in case–control studies, the OR is a commonly used statistic for measure of association and risk assessment. In the current study, we used the logistic regression method entering sex as a covariate and the additive method developed by Clayton6 to reanalyse the X chromosome data from our GWAS for Graves’ disease; the two methods gave consistent results, both showing that GPR174 was associated with disease susceptibility.
GPR174 consists of one exon encoding a protein of 333 amino acids, which belongs to the GPCR superfamily and is grouped into GPCR 1 (or rhodopsin-like) family. These integral membrane proteins are characterised by the presence of seven α-helical transmembrane domains and play important roles in cell signal transduction. To date, more than 50% of the effective drug targets are GPCRs.26 ,27 Very recently, lysophosphatidylserine (LysoPS) was found as a ligand for GPR174.28 ,29 LysoPS is secreted by the immune system in vivo, and acts a lipid mediator that regulates immune system processes.29 LysoPS interacting with GPR174 stimulates an increase of intracellular cyclic adenosine monophosphate (cAMP) in a dose dependent manner.29 cAMP-elevating or cAMP-mimicking agents could inhibit production of the T-helper 1 (Th1) cytokines, whereas production of the Th2 cytokines remains unchanged or even enhanced.30 It is often considered that Graves’ disease is a Th2 disorder. Therefore, the elevated level of cAMP might drive the Th2 polarisation and be involved in the pathogenesis of Graves’ disease. This assumption corresponded with our result that the susceptible T allele of rs3827440 was associated with a higher level of GPR174 expression, since the higher level of GPR174 expression might elevate the intracellular cAMP concentration. Our RT-PCR analysis showed GPR174 was widely expressed in immune related tissues and moderately expressed in thyroid. This expression pattern suggested a possible involvement of GPR174 in immune processes and a potential link of this gene to the thyroid structure/function, which could be crucial in the aetiology of Graves’ disease. Graves’ disease shares genetic susceptibility factors with other autoimmune diseases such as type 1 diabetes, multiple sclerosis, and rheumatoid arthritis.4 It is therefore reasonable to address the possibility of an association of GPR174 with these autoimmune diseases in the future.
XCI skewing or other X chromosome associated abnormalities might contribute to disturbances in self reaction and ultimately to autoimmunity, which might enhance the susceptibility of the female sex to autoimmune disease including Graves’ disease. Several hypotheses have been proposed to explain the mechanisms.31 The loss of mosaicism hypothesis postulated that autoreactive T cells may fail to be tolerated by self-antigens encoded by one of the two X chromosomes, and these autoreactive T cells may stimulate B cells and induce autoimmunity in the periphery.32 A high prevalence of skewed XCI in females with Graves’ disease supports this hypothesis.33–35 The haploinsufficiency hypothesis states that haploinsufficiency for X-linked genes results in some autoimmune disorders.31 ,36 The observation that women with Turner's syndrome (loss of one X chromosome; X0) have an increased risk of developing autoimmune thyroid disease including Graves’ disease corroborated this hypothesis.37 Other evidence supporting this hypothesis is the higher rate of circulating cells with X chromosome monosomy that were found in females with Graves’ disease and other autoimmune disorders.36 Further study is required to clarify whether GPR174 is involved in the X chromosome-specific abnormalities or whether GPR174 plays a role in the pathogenesis of Graves’ disease via the above mechanisms.
Although skewing of XCI or X chromosome associated abnormalities might play a role in female preponderance, it is unlikely that variation in a single gene would be responsible for the higher susceptibility of females to Graves’ disease. If this was the case, we would expect a higher frequency of autoimmune diseases in males. The risks in males and females derived from genotype distribution of rs3827440 (table 1) support minimal female:male difference in risk. Specifically, using the larger replication cohort, we obtained relative risks (RRs) of 1.60 for the T allele in males, and RRs of 1.68 and 1.48 for the TT and TC genotypes in females, respectively. These RRs gave an attributable risk of 26% in males and 31% in females, attributable to this polymorphism—that is, nearly no difference. In fact, using both the original and replication data together provides even less difference. Therefore, the genotype distributions of rs3827440 explain little of the difference in Graves’ disease prevalence between males and females in the Chinese population.
In conclusion, the identification of GPR174 as a risk factor for Graves’ disease resulted from an extended analysis of the X chromosome data. Exploring the X chromosome data in GWAS studies more carefully and making full use of the sample would help to reveal the X-linked loci with susceptibility to complex disease. This study has not only identified a new X chromosome risk locus for Graves’ disease but also suggests the X chromosome targets for study in other autoimmune diseases.
Useful websites
-
snpMatrix, http://bioc.ism.ac.jp/2.8/bioc/html/snpMatrix.html
-
R statistical environment V.2.13.2, http://www.r-project.org/
-
PLINK v1.07, http://pngu.mgh.harvard.edu/~purcell/plink/
-
IMPUTE2, http://mathgen.stats.ox.ac.uk/impute/impute_v2.html
▸ SNPTEST v2, https://mathgen.stats.ox.ac.uk/genetics_software/snptest/snptest.html.
Acknowledgments
We thank all subjects for participating in this study. We gratefully acknowledge Dr Ethan Lange for his valuable comments and kind help in improving the quality of the manuscript. This work was supported in part by the Public Benefit Research Foundation from Ministry of Health of China (201202008), National Natural Science Foundation of China (31271343, 31171222 and 31000556), funds from Shanghai Science and Technology Committee (11DJ1400204, 10ZR1421400) and Shanghai Rising-Star Program (12QA1402400).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors WH, ZC, S-JC and XC designed the research; WH, XC and ZC wrote the paper; XC, WH, X-JM and Y-NB analysed the data; XC, MS, FX, W-HS, K-YZ, LY P-PY, QH, H-FW and J-XS performed the research; LL, W-DL, YD, YW and H-DS collected clinical samples.
-
Funding Public Benefit Research Foundation from Ministry of Health of China (201202008), National Natural Science Foundation of China (31271343, 31171222 and 31000556), Shanghai Science and Technology Committee (11DJ1400204, 10ZR1421400), Shanghai Rising-Star Program (12QA1402400).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The Ethical Committee of the Chinese National Human Genome Center at Shanghai.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/