Article Text

Abstract
Background Here we have developed a novel and much more efficient strategy for the complete molecular characterisation of the cystic fibrosis (CF) transmembrane regulator (CFTR) gene, based on multiplexed targeted resequencing. We have tested this approach in a cohort of 92 samples with previously characterised CFTR mutations and polymorphisms.
Methods After enrichment of the pooled barcoded DNA libraries with a custom NimbleGen SeqCap EZ Choice array (Roche) and sequencing with a HiSeq2000 (Illumina) sequencer, we applied several bioinformatics tools to call mutations and polymorphisms in CFTR.
Results The combination of several bioinformatics tools allowed us to detect all known pathogenic variants (point mutations, short insertions/deletions, and large genomic rearrangements) and polymorphisms (including the poly-T and poly-thymidine-guanine polymorphic tracts) in the 92 samples. In addition, we report the precise characterisation of the breakpoints of seven genomic rearrangements in CFTR, including those of a novel deletion of exon 22 and a complex 85 kb inversion which includes two large deletions affecting exons 4–8 and 12–21, respectively.
Conclusions This work is a proof-of-principle that targeted resequencing is an accurate and cost-effective approach for the genetic testing of CF and CFTR-related disorders (ie, male infertility) amenable to the routine clinical practice, and ready to substitute classical molecular methods in medical genetics.
- Cystic fibrosis
- Diagnostics
- Molecular genetics
- Genetic screening/counselling
- Getting Research into Practice
Statistics from Altmetric.com
- Cystic fibrosis
- Diagnostics
- Molecular genetics
- Genetic screening/counselling
- Getting Research into Practice
Introduction
Cystic fibrosis (CF; MIM #219700) is one of the most common, life-threatening, autosomal recessive genetic disorders, with a carrier frequency in the Caucasian population of around 1 in 20–80 people.1 Mutations in the CF transmembrane conductance regulator (CFTR/ABCC7; MIM #602421) gene determine the impairment of chloride transport in epithelial cells, mainly affecting lungs, digestive tract, sweat glands and vas deferens in men.2 Although a major mutation (deltaF508) accounts for over two-thirds of CF alleles worldwide,3 a high level of allelic heterogeneity has been described within different CF populations,4 including single nucleotide variants (SNVs), short insertions and deletions (InDels) and large structural variants (SVs). Since the characterisation of CFTR more than 20 years ago,5–7 1937 CFTR variants have been reported (Cystic Fibrosis Mutation Database, http://www.genet.sickkids.on.ca). In addition to the classical CF phenotype, mild mutations in CFTR can cause other CFTR-related disorders (CFTR-RD), such as male infertility due to congenital bilateral absence of the vas deferens (CBAVD; MIM #277180), idiopathic chronic pancreatitis (MIM #167800), and bronchiectasis (MIM #211400) among others.8 Some of these mild alleles are common polymorphisms, such as poly-thymidine (poly-T) and poly-thymidine-guanine (poly-TG) tracts, associated with aberrant splicing of exon 10 of CFTR, being the most common mutation in CBAVD.9 Although CFTR is one of the most extensively studied human disease genes, its high allelic heterogeneity makes CF and CFTR-RD molecular diagnostics challenging.
The precise diagnosis of CF combines clinical evaluation (clinical features of CF phenotype and sweat test measurements) with CFTR molecular genetic studies. To date, the molecular characterisation of CFTR mutations in a given sample relies on commercial tests that screen for specific common mutations (reverse dot blot INNO-LIPA CFTR [Innogenetics], Cystic Fibrosis Genotyping Assay/OLA [Abbott], Elucigene CF-EU2 [Zeneca], xTAG Cystic Fibrosis 71 kit v2 [Luminex], among others). The detection rate of these panels varies depending on the mutations included (ranging from 4 to 70 CFTR mutations) and the molecular heterogeneity of each population. For many patients with common CFTR mutations that are present in these commercial panels, there is no need for additional studies, but the high heterogeneity of CFTR mutations in some CF populations and in CFTR-RD, often makes necessary the complete molecular screening of the 27 exons and the regulatory regions of CFTR, which is a costly and labour-intensive task.
As a first step towards the implementation of next-generation sequencing (NGS) approaches to molecular testing that can replace current low-throughput and time-consuming molecular methods, we assessed the efficacy of targeted resequencing for the molecular diagnosis of CF and CFTR-RD in a heterogeneous panel of 92 patients with CF and CFTR-RD, and CF carriers with known CFTR mutations.
Materials and methods
Detailed protocols are available in online supplementary materials.
Subjects
High-quality genomic DNA from 92 unrelated samples, including patients with CF (n=45), CF carriers (n=27) and patients with CFTR-RD (n=20), were extracted from peripheral blood lymphocytes using standard protocols. The group of subjects with CFTR-RD included 12 patients with CBAVD, 5 patients with idiopathic bronchiectasis, and 3 patients with CFTR-related metabolic syndrome. All samples included in this study had previously undergone conventional CFTR screening,10 ,11 and all CFTR mutations were confirmed by Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA) or quantitative PCR (qPCR). The samples selected for this study were recruited for diagnostic purposes between 1998 and 2011. For obvious reasons, it has been impossible to obtain the corresponding informed consents, although all samples were obtained with the purpose of CFTR mutation screening. For that, all samples were anonymised in order to ensure the protection of their identity and the list of confirmed mutations was not provided to the investigators performing the bioinformatics mutation analysis until the end of the variant prioritisation process.
Insolution capture and multiplexed resequencing of CFTR
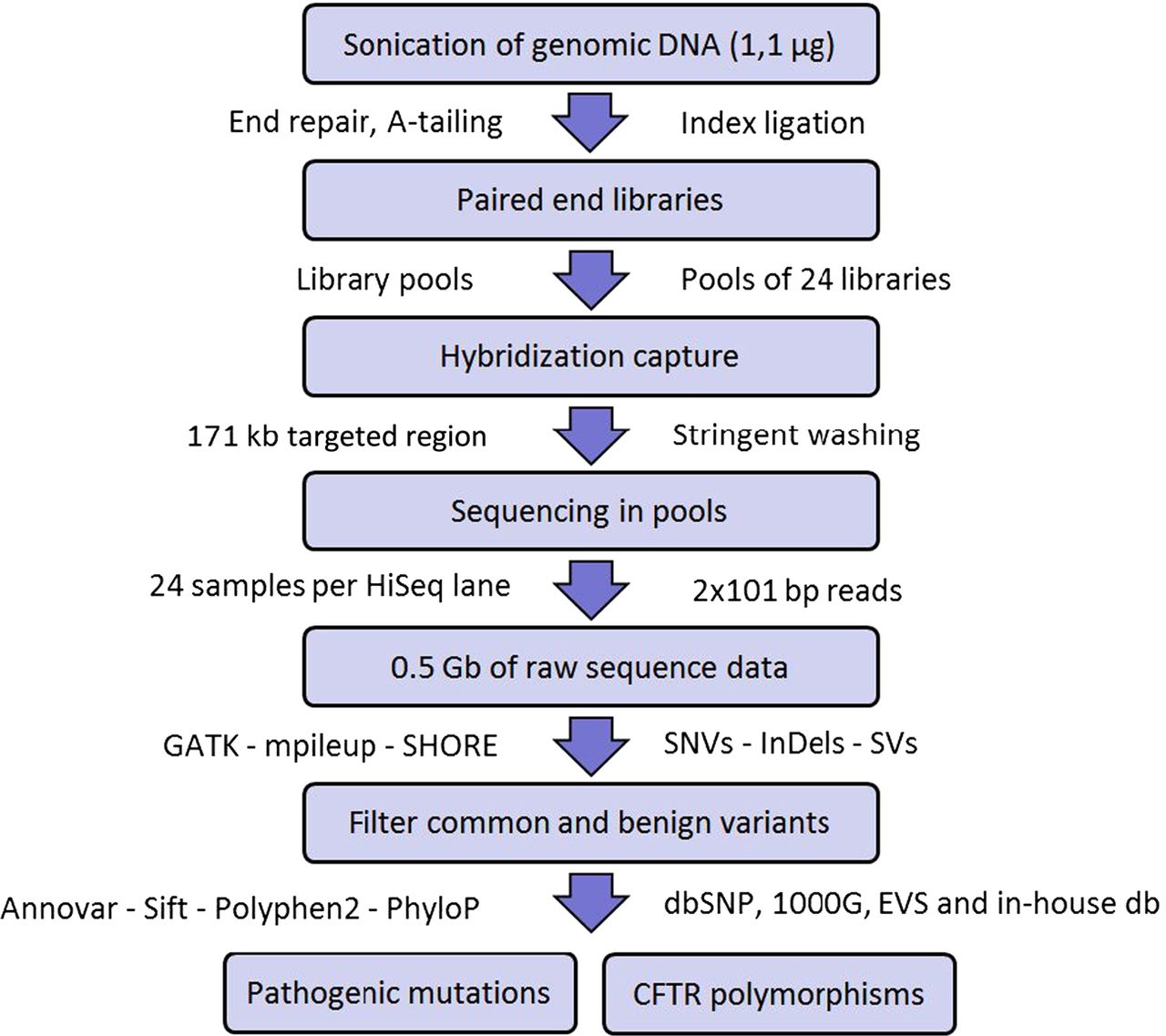
Figure 1 summarises the mutation screening workflow that we have implemented in this study. Briefly, DNA from blood was sonicated to obtain fragments of approximately 200 bp. Then, fragments underwent end repair, A-tailing, and ligation to Illumina paired-end indexed adapters, as outlined in the DNA Truseq protocol (Illumina). Once the DNA libraries were indexed, they were PCR amplified and pooled before in-solution hybridisation to a custom NimbleGen SeqCap EZ Choice Library (Roche) of CFTR complementary oligonucleotide DNA baits. After stringent washing, the captured libraries were PCR amplified and sent for sequencing (24 libraries per lane) to generate 2×101 bp paired-end reads with a HiSeq 2000 instrument (Illumina). Finally, the resulting DNA sequences were aligned to the human reference genome and sequence variants were detected and annotated as outlined in online supplementary materials.

Assay workflow to identify CFTR polymorphisms and pathogenic mutations.
Identification of CF and CFTR-RD mutations
In order to identify CFTR pathogenic mutations that could cause CF and CFTR-RD, we applied the following filtering steps12:
-
We required all candidate variants on both sequenced DNA strands and to account for ≥15% of total reads at that site.
-
Common polymorphisms (≥5% in the general population) were discarded by comparison with National Center for Biotechnology Information (NCBI) single nucleotide polymorphism (SNP) Database (dbSNP) build 132, the March 2010 release of the 1000 Genomes project (http://www.1000genomes.org), the Exome Variant Server (http://evs.gs.washington.edu) and an inhouse exome variant database to filter out common benign variants and recurrent artefact variant calls. However, since these databases contain known disease-associated mutations, all detected variants were compared with gene-specific mutation databases (http://www.hgmd.cf.ac.uk and http://www.genet.sickkids.on.ca).
-
Then, we screened for mutations that could give rise to premature protein truncating mutations, that is, stop mutations, damaging missense variants, splice sites, exonic deletions/insertions and large SVs.
-
Variants were ranked based upon evolutionary conservation and potential deleteriousness of the affected nucleotide using Sift,13 Polyphen2,14 PhyloP,15 and MutationTaster.16
Results
CFTR enrichment
We designed oligonucleotides to target the complete genomic sequence (the 27 exons plus all introns), and 10 kb of 5′ and 3′ flanking genomic regions of CFTR covering a total of 208 701 bp. After removal of repetitive sequences, 87% of the targeted bases could be covered with capture baits for a total targeted region of 181 539 bp in 171 individual regions, with lengths ranging from 68 bp to 6689 bp (average 1062 bp). We included the untranslated region of CFTR to have a complete definition of the non-coding variability and to favour the detection and sizing of large SVs within the gene.
CFTR sequencing statistics
On average, for each of the four HiSeq2000 (Illumina) lanes, 95.8% of the paired-end 2×101 bp reads could be assigned unambiguously to individual samples, according to their tags, receiving similar proportion of reads for each sample. The remaining 4.2% of unassigned reads were removed because of sequencing errors in their index tags. Therefore, the losses of sequence data associated with high sample multiplexing were minimal. On average, for every sample, 95% of high quality sequencing reads mapped to the reference genome. This resulted in an evenly distributed mean depth of coverage for CFTR of 231X (199X if the targeted regions are expanded by 150 bp at each end) with a coefficient of variation of 35%, across samples. In fact, 99.7% of all targeted bases were covered by at least 5 reads (the minimum that we require for variant calling) and 78.53% by at least 100 reads (table 1). For a comprehensive summary of the obtained sequencing results, see also online supplementary table S1.
Sequencing quality control parameters, coverage and detected variants by targeted resequencing of the CFTR gene using pools of 8, 12, 16 and 24 samples
To determine if coverage was substantially lower for any region, we calculated the proportion of base pairs that were captured by <50 reads. The proportion of these poorly covered regions accounted for 0.069 of CFTR targeted bases, and only 0.07% of the targeted bases were not covered by any read (table 1). As expected, these low-covered genomic regions are characterised by low complexity and a high GC content. Sequence targets with these two characteristics are usually refractory to enrichment, resulting in reduced coverage for these sites. However, as shown above, this was the case for only a very small proportion of all bases intended to be captured in this study. From these data we can conclude that all samples, regardless of the pool sizes in the precapture step, were uniformly covered at depths that in all cases exceed by far the minimum coverage required for a reliable variant calling (see online supplementary table S1). The minor differences between samples and pools were neutralised by the excessive overall CFTR coverage achieved by our assay. The sequence quality metrics of this data warrant a confident detection of variants in all samples.
Identification of CF and CFTR-RD mutations
The selection of the samples for this study was done with the idea to include as many different types of CFTR mutations as possible, to simulate a real-world diagnostics scenario, including SNVs, InDels, and large SVs, so that we could test the performance of our approach for all these types of genetic variations. To assess the sensitivity of our assay to detect pathogenic mutations, we blindly inspected all mapped sequence reads from the 92 samples with previously defined mutations in CFTR.
By using our multiplexed capture approach and automated variant calling pipeline, we were able to detect, before variant filtering and ranking, 115 SNVs (4 novel) and 28 InDels (19 novel) in CFTR per sample on average (table 1). Among these variants we identified several common CFTR polymorphisms (see online supplementary table S2). Then, we applied our variant prioritisation strategy to identify CFTR pathogenic mutations present in each sample. Using this strategy we detected 122 different pathogenic mutations on CFTR in their correct heterozygous/homozygous state across the 92 samples included in the study (some variants were present in more than one sample). We correctly identified 58 missense, 14 nonsense, 23 splice site SNVs, 12 frameshift deletions, 2 frameshift insertions, and 3 inframe deletions (one of 84 nucleotides long), known to cause CF and CFTR-RD (see online supplementary table S3). In addition, we were also able to detect three different 5T pathogenic haplotypes, five large deletions, one duplication and one large genomic rearrangement (that includes one inversion and two deletions) involving various CFTR exons.
Intron 9 poly-TG and poly-T haplotypes and alternative splicing of CFTR
The 5T variant in intron 9 (c.1210–12T[5] is the most common mutation associated with CBAVD.9 The penetrance of the 5T variant depends on the neighbouring TG sequence repeat.17 Thus, the definition of the TG-T (c.1210–34TG[11–13]T[5–9]) haplotype contributes to predict the most likely CFTR-RD phenotype of the carrier subject. However, the repetitiveness of its sequence at the nucleotide level makes difficult to determine the TG-T haplotype using standard variant calling algorithms (figure 2). In order to address this issue, we developed an in-house script that scans the very raw sequencing data of each sample for all possible combinations of c.1210-34TG[11–13]T[5–9]. By doing this, we were able to determine the exact TG-T haplotype of each sample, including three T5-TG11, eight T5-TG12 and two T5-TG13 haplotypes (see online supplementary table S4).
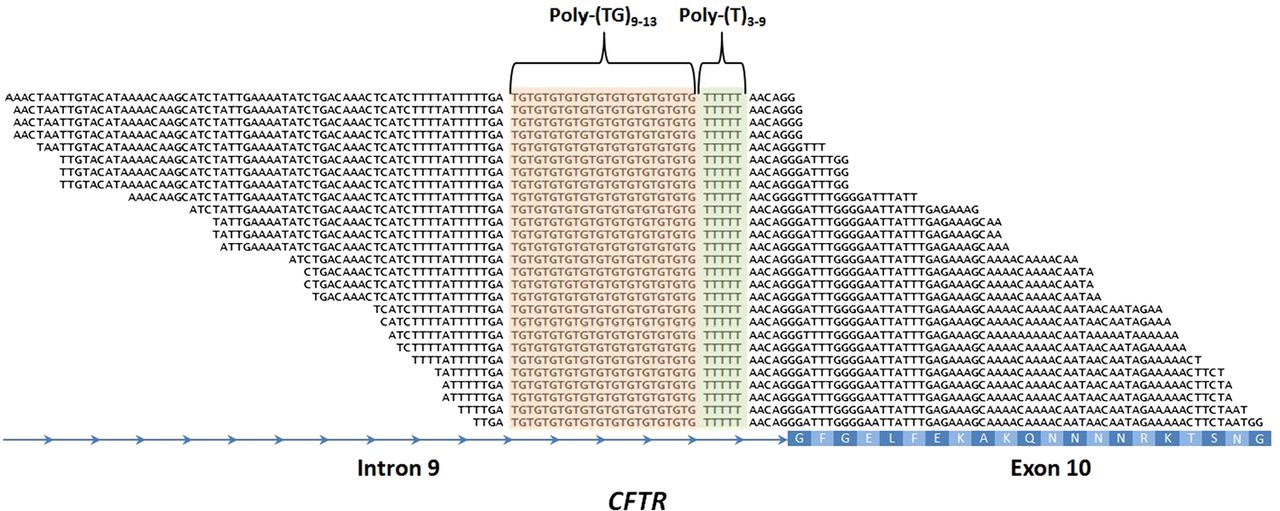
Detection of the intron 9 poly-TG-T haplotype involved in male infertility and other CFTR-RD. Example of a patient with CFTR-RD with the c.1210-34TG[12]T[5] haplotype. The centre of the alignment of the 100 nt NGS reads shows the poly-TG (in orange) and poly-T (in green) tracts. The CFTR intron 9 and exon 10 (with the amino acid sequence in white) are represented in the bottom in blue. poly-TG, poly-thymidine-guanine.
Characterisation of large structural changes in CFTR
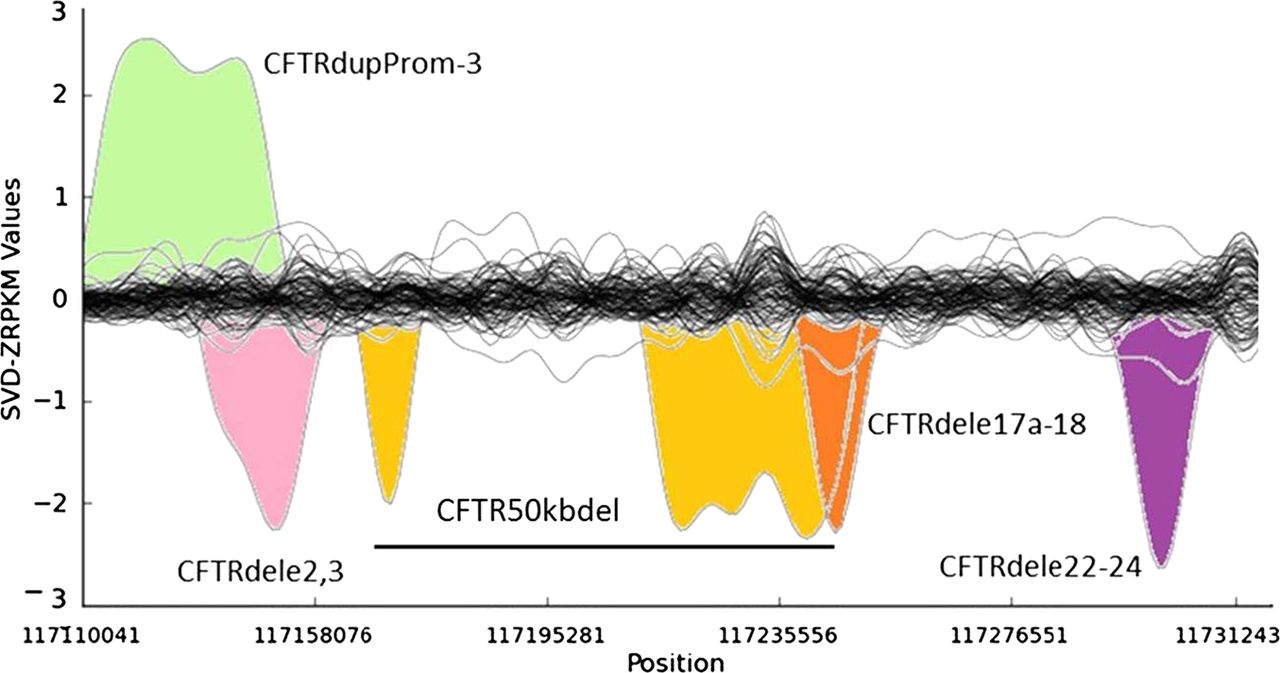
Several of the unknown CF and CFTR-RD mutations in affected individuals may not have been identified yet because of the intrinsic low sensitivity of traditional PCR-based CFTR screening approaches for large SVs. It has been estimated that large genomic rearrangements of CFTR, which exhibit extensive allelic heterogeneity and are mainly caused by non-homologous recombination events, may account for up to 20% of the unidentified CFTR alleles in patients with CF and CFTR-RD.18 A major step-forward of NGS technologies with respect to classical molecular approaches is the possibility to detect large genomic rearrangements at the same time than SNVs and InDels, without the need for additional assays specific for large SVs, such as array-comparative genomic hybridisation, semi qPCR based methods, MLPA or quantitative multiplex PCR of short fluorescent fragments. In our study, the combination of paired-end mapping, split-read analysis, and normalised depth of coverage strategies allowed the blind identification of 7/7 (100% sensitivity) large SVs (5 deletions, one duplication and one complex rearrangement) in CFTR (figure 3). We were able to accurately identify the breakpoints of all of them, with a perfect concordance between the prediction of the algorithms and the validations for each of them (table 2).
Large structural variants identified in the CFTR by targeted resequencing
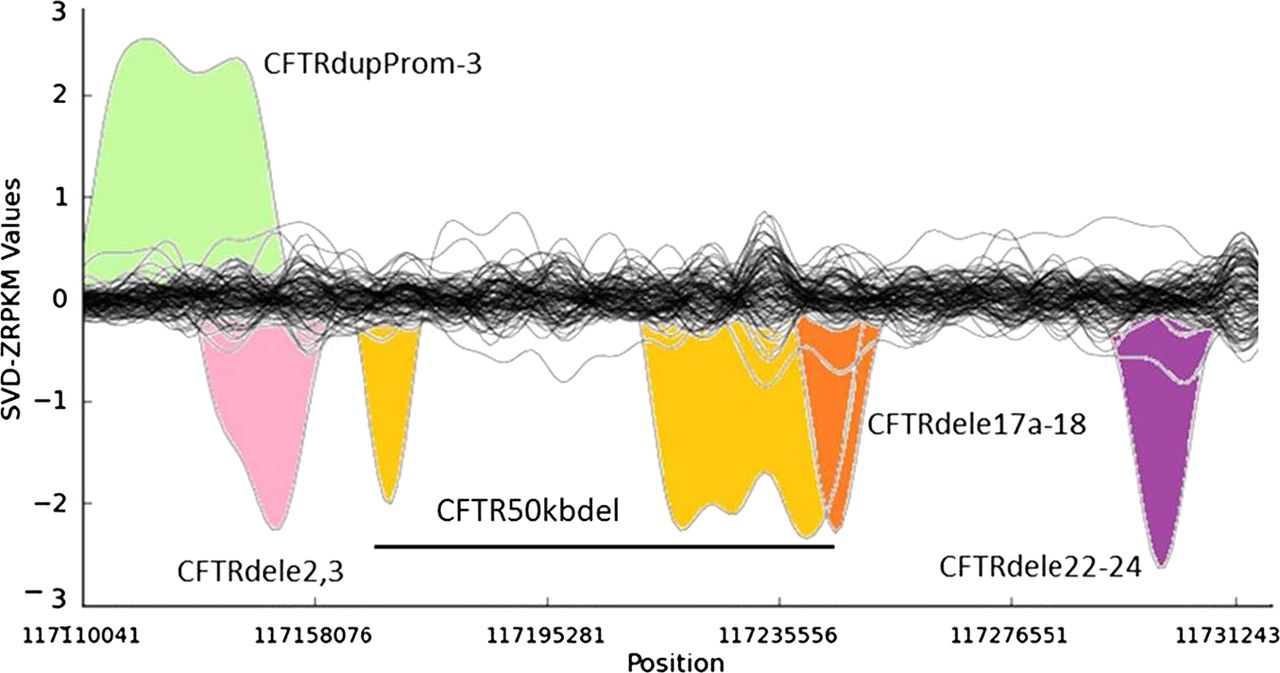
Detection of large structural variants in the CFTR gene by normalised depth of coverage analysis. Representation of the SVD-ZRPKM Values calculated by Conifer29 for the 92 samples. Coloured peaks indicate the five largest structural variants identified in this study.
Among the seven SVs analysed in this study we have also characterised in silico and validated by Sanger sequencing the breakpoints of a novel (ie, not previously reported to the public databases) CFTR 1899 bp deletion (chr7:117267155-117269054, hg19) that includes the loss of exon 22 (c.3469-420_3717+1230del1899), and all the breakpoints of a large genomic rearrangement previously reported as CFTR50kbdel (legacy name).21 These two SVs were previously identified in their respective samples by means of MLPA and qPCR, but their breakpoints were not known. Thanks to the results of this study now we know that CFTR50kbdel consists of a 85 kb inversion, with breakpoints chr7:117169862/117169876-117255003; containing two large deletions: chr7:117169908-117180511(10.6 kb deletion of exons 4–8), and chr7:117216401-117254987(38.6 kb deletion of exons 12–21), 49.2 kb in total, which is remarkably close to the 50 kb deletion originally estimated by classical molecular methods21 (figure 4A,B). In addition, cDNA analysis evidenced an aberrant transcript showing a unique junction exon 3/22 indicating the loss of the entire inverted region (figure 4C). This is the first time that a CFTR large inversion is reported, and, to our knowledge, it is the most complex rearrangement ever characterised in CFTR (c.[274-1091_3468+236inv85141ins38; 274-1044_1116+111del10602insTATAT; 1585-11 392_3468+219del38585]).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the complex CFTR50kbdel. (A) Normal structure of CFTR. Black boxes represent each of the 27 CFTR exons. (B) Diagram shows the complex architecture of the CFTR50kbdel mutation. Arrows indicate the breakpoints of the 85 kb inversion. Grey areas indicate the two deleted regions. (C) cDNA sequence of CFTR50kbdel, showing the loss of exons 4–21.
Sensitivity and specificity of targeted resequencing of CFTR
The molecular diagnostic strategy for CF and CFTR-RD that we present here has blindly identified all previously known pathogenic CFTR variants in the 92 CF samples studied. This represents a mutation detection rate of 100% (122/122), with zero false-positive calls, and would have resulted in a positive molecular diagnosis in 91 of the 92 patients with CF and CFTR-RD and CF carriers (diagnostic rate of 98.9%), since for one of the patients with CF (sample 80) we were unable to identify his previously unknown second CF allele. As expected, for patients with CFTR-RD with only one previously known CFTR mutation our NGS strategy hasn't identified a second CFTR allele, as it is the case of three idiopathic bronchiectasis and four patients with CBAVD. It is known that other genetic and environmental factors may contribute to these phenotypes,23–25 so the apparently missing CFTR alleles in these samples cannot be solely attributed to issues with the specificity or sensibility of our approach. Overall, the high success rate achieved in this study highlights the accuracy of this strategy as a molecular diagnostics tool for CF and CFTR-RD.
Precapture pooling and multiplexed sequencing reproducibility
Precapture pooling reduces substantially the library preparation time and, in combination with multiplexed sequencing, allows to exploit the full potential of NGS for clinical diagnostics. In order to assess how precapture multiplexing affects coverage and accuracy, we tested different pool sizes: two captures of 8, 12 and 16 samples each and one capture of 24 samples. All samples were marked with a specific index/tag, so that their individual identification was warranted at the end of the sequencing run. The sequence quality data and the variant calling results indicate that there were no sensitivity or specificity problems associated with the use of precapture pools of high number of samples (table 1). Thus, the major technical consequences of precapture pooling, which are the reduction in the input amount of the individual libraries and the addition of multiple barcodes, which may lead to less efficient blocking and favour unspecific hybridisation,26 have minor effects in the final variant calling process.
Reproducibility was determined by running four samples (two patients with CF and two patients with CFTR-RD) in duplicate on the same run, but captured in pools of different sample sizes (in two different precapture pools of 8 and 24 samples, respectively), and sequenced in independent HiSeq2000 lanes. We detected eight of eight pathogenic mutations in the replicated samples, yielding 100% reproducibility of mutation detection. We next assessed the reproducibility for all variant calls in the entire CFTR captured region (mean=138±47 SNVs and 32±11 InDels per sample). Across the four samples, reproducibility was 96.09% for SNVs and 71.62% for InDels, with an overall reproducibility of 91% for all variants in all four samples (table 3). Variant calls that did not replicate were all intronic or in intergenic regions, and almost all of them were located very close to the ends of the targeted regions of CFTR or in regions not covered by capture baits. This explains the observed low coverage of these unreplicated variants (mean=33.47X vs 144.58X of the replicated variants), and highlights the impact of the depth of coverage on the assay reproducibility.
Assay reproducibility of the identification of CFTR mutations by targeted resequencing
Twenty-one out of the 122 pathogenic variants detected by our analysis were present in two or more individuals (see online supplementary table S3). This means a reproducibility of 100% for pathogenic variant calls between two or more samples (based on the results of 17.21% of the mutations included in this study). Since most of the samples bearing these mutations were multiplexed in independent precapture pools of different sample sizes, and also were run in different sequencer lanes, we can conclude that our approach offers great robustness and reproducibility in the detection of CFTR pathogenic variants. Although the coverage for a given mutation can vary significantly between samples, the proportion of reads supporting the non-reference allele was always maintained (see online supplementary table S3). Altogether, these results highlight the sensitivity and reproducibility of our assay, and support the use of a larger number of samples in precapture pools in future studies, when more index tags are available (24 when we planned this study).
Discussion
Here we have implemented and tested a novel strategy for the molecular analysis of CF and CFTR-RD, based on pooled target enrichment and multiplexed NGS of CFTR. We have validated this new approach in a cohort of 92 samples with previously known pathogenic CFTR mutations. The different pools of simultaneously enriched CFTR samples were multiplexed in groups of 24 samples in four sequencer lanes. After mapping the sequencing reads to the reference genome and performing blind variant calling and filtering, our bioinformatics pipeline successfully retrieved all known pathogenic mutations in their correct heterozygous/homozygous state. With this approach we were able to identify a heterogeneous panel of CFTR mutations, including SNVs, InDels and large SVs. Our results (mutation detection rate of 100% and diagnostic rate of 98.91%) demonstrate the suitability of targeted resequencing for the routine clinical diagnosis of CF and CFTR-RD.
Clinical diagnostic tools must meet very stringent sensitivity and specificity parameters, while keeping their cost-effectiveness and time-effectiveness. The approach that we describe here represents a change in the paradigm for the molecular diagnostics of CF and CFTR-RD. Until now, the ideal strategy for CFTR screening consisted of three sequential steps:10 (1) genotyping by commercially available kits a small subset (30–50) of common CFTR mutations; (2) in case of not having identified the two CFTR alleles, complete screening of the coding portion and flanking regions of CFTR by scanning techniques, like denaturing gradient gel electrophoresis or single strand conformation polymorphism/heteroduplex among others, and subsequent Sanger sequencing; and if still insufficient, (3) screening by MLPA and/or array-comparative genomic hybridisation for large genomic rearrangements. The average cost per sample of this strategy is around €400 with an estimated turnaround time of 2–3 months for samples that have to undergo all three steps described above. We estimate that the approach that we present here has an overall cost of less than €200 per sample, which represents a 50% of cost savings per sample and makes the whole process eight times faster when compared with the techniques currently used for the molecular diagnosis of CF and CFTR-RD. In addition, our strategy offers a complete definition of the captured CFTR, without the need for stepwise testing anymore. We foresee that these differences will become even more significant because of the constantly dropping sequencing costs27 and optimised library preparation and sequencing protocols. The complete process of library preparation, sequence enrichment, NGS and bioinformatics analysis could be completed within 14 days after reception of the DNA sample. The most time-consuming step was sequencing the CFTR-enriched DNA libraries on the HiSeq2000 (Illumina), which took approximately 10 days. In addition to saving time in the process of library preparation with new capture strategies, using the most recent enrichment technologies such as Haloplex (Agilent), and optimising the bioinformatics, major time savings could be made by using the new generation of HiSeqs (Illumina) sequencers (series 2500), which have been recently reported to be able to generate up to 140 GB of sequence (2×100 bp) in approximately 24 h.28 As an alternative that would reduce NGS costs, we propose the use of smaller, benchtop, personal sequencers such as the MiSeq (Illumina) or Ion Torrent (Ion Torrent Systems). The amount of sequence output of these instruments is approximately 10 times smaller than its bigger siblings, so they would be ideal for the analysis of batches of reduced numbers of samples (up to 10 samples per run).
The major drawback of capturing the complete genomic sequence of CFTR instead of focusing only on the coding regions is that more sequencing is needed to achieve similar coverage. However, the benefits of this approach are that no deep intronic mutations are missed, nor variants in the promoters or in the Untranslated regions (UTRs). In addition, this strategy has also proven its utility to detect large deletions, duplications and inversions, involving various CFTR exons, as well as to detect their breakpoints. The detection of variation in the untranslated regions of CFTR can also be used for the identification of alleles of clinical relevance, such as the 5T variant, which has variable penetrance and accounts for part of the phenotypic variability of CFTR-RD.17
In addition to the technical limitations inherent to hybrid capture, such as selection bias and uneven capture efficiency, the main limitation of the targeted resequencing approach is the impossibility to efficiently capture and sequence the repetitive and low-complexity, and GC-rich genomic segments of CFTR that are refractory to enrichment. However, the constant optimisation of the capture probes and NGS chemistries will gradually close the capture gaps (mainly due to uniqueness constraints, homopolymer runs, ambiguous bases or other factors that are known to cause issues in either oligonucleotide synthesis or hybridisation), and reduce enrichment variability between samples. But until then, this will require backup methods to assess the variability in these ‘dark’ regions, in the case of samples with clear CF or CFTR-RD phenotypes, but with no identified mutations in the captured fraction of CFTR, as in the case of sample 80 for which we were not able to find its previously unknown second CF allele. However, the major sources variability that could potentially affect the sensitivity and specificity in our study (such as variations in Guanine-cytosine (GC) content or differential hybridisation efficiency of the two alleles in a diploid genome) are neutralised by the high level of sequencing depth achieved.
The transition of NGS technologies from basic research to routine molecular diagnostics over the next years, will take advantage of the constant improvements in the reliability and robustness of these technologies, and of simplified bioinformatics analyses able to generate medical report-like outputs adapted to clinical laboratories. We are still in the process of defining the methods and guidelines for the application of NGS to clinical genetic diagnostics. In this initial phase, we still recommend that novel mutations are validated by Sanger sequencing before informing the patient.
In conclusion, this represents, to the best of our knowledge, the first study successfully using targeted NGS to detect pathogenic lesions in the CFTR gene. With the approach reported here we have been able to describe for the first time the breakpoints of a novel deletion and the most complex genomic rearrangement in CFTR. We have only had one false positive and zero spurious calls. Altogether, our assay shows a clear superiority with respect to traditional methods for CFTR screening and overcomes their technical limitations, making it their natural replacement in the diagnostic laboratories.
Acknowledgments
We thank the subjects and referring physicians who participated in this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors This study was conceived and designed by DT, TC, LA, and XE. Selection of samples was performed by TC. NGS libraries were prepared by JG. The bioinformatics pipeline and the NGS analysis was performed by DT, CT, GE, and SO. Validation of SVs was performed by FS, MDR, and TC. The manuscript was written by DT, TC, and XE. All aspects of the study were supervised by TC and XE.
-
Funding This project was funded by the Spanish Plan Nacional SAF2008–00357 (NOVADIS); the Generalitat de Catalunya AGAUR 2009 SGR-1502; the Instituto de Salud Carlos III (FIS/FEDER PI11/00733); and the European Commission 7th Framework Program, Project N. 261123 (GEUVADIS), and Project N. 262055 (ESGI). DT is a PhD student supported by the Spanish Ministry of Economy and Competiveness; FS is a PhD student from Associació Catalana de Fibrosi Quística.
-
Competing interests None.
-
Ethics approval PRBB Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.