Article Text

Abstract
Background Congenital nephrotic syndrome arises from a defect in the glomerular filtration barrier that permits the unrestricted passage of protein across the barrier, resulting in proteinuria, hypoalbuminaemia, and severe oedema. While most cases are due to mutations in one of five genes, in up to 15% of cases, a genetic cause is not identified. We investigated two sisters with a presumed recessive form of congenital nephrotic syndrome.
Methods and results Whole exome sequencing identified five genes with diallelic mutations that were shared by the sisters, and Sanger sequencing revealed that ARHGDIA that encodes Rho GDP (guanosine diphosphate) dissociation inhibitor α (RhoGDIα, OMIM 601925) was the most likely candidate. Mice with targeted inactivation of ARHGDIA are known to develop severe proteinuria and nephrotic syndrome, therefore this gene was pursued in functional studies. The sisters harbour a homozygous in-frame deletion that is predicted to remove a highly conserved aspartic acid residue within the interface where the protein, RhoGDIα, interacts with the Rho family of small GTPases (c.553_555del(p.Asp185del)). Rho-GTPases are critical regulators of the actin cytoskeleton and when bound to RhoGDIα, they are sequestered in an inactive, cytosolic pool. In the mouse kidney, RhoGDIα was highly expressed in podocytes, a critical cell within the glomerular filtration barrier. When transfected in HEK293T cells, the mutant RhoGDIα was unable to bind to the Rho-GTPases, RhoA, Rac1, and Cdc42, unlike the wild-type construct. When RhoGDIα was knocked down in podocytes, RhoA, Rac1, and Cdc42 were hyperactivated and podocyte motility was impaired. The proband's fibroblasts demonstrated mislocalisation of RhoGDIα to the nucleus, hyperactivation of the three Rho-GTPases, and impaired cell motility, suggesting that the in-frame deletion leads to a loss of function.
Conclusions Mutations in ARHGDIA need to be considered in the aetiology of heritable forms of nephrotic syndrome.
- Renal Medicine
- Cell Biology
- Clinical Genetics
- Developmental
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Congenital nephrotic syndrome is a rare kidney disorder characterised by the presence of massive proteinuria, hypoalbuminaemia, and generalised oedema in the first 3 months of life. It arises from a defect in the glomerular filtration barrier that permits the unrestricted passage of protein across the barrier into the urine. The glomerular filtration barrier consists of fenestrated endothelial cells, the acellular glomerular basement membrane that is about 300 nm thick, and specialised epithelial cells called podocytes. The majority of children with congenital nephrotic syndrome have a monogenic basis for their disease due to mutations in genes that affect the structure and function of the actin cytoskeleton within podocytes.1 In a large survey, ∼85% of cases of congenital nephrotic syndrome were attributed to mutations in NPHS1 (nephrin), NPHS2 (podocin), LAMB2 (laminin β2), or WT1 (Wilms tumour suppressor 1), while the remaining 15% were genetically uncharacterised.1 Subsequent to this report, mutations in PLCE1 (phospholipase C ɛ) were also identified in cases of congenital nephrotic syndrome, more frequently in the presence of a renal biopsy showing diffuse mesangial sclerosis.2 ,3 Establishing a genetic diagnosis has important implications for the treatment of congenital nephrotic syndrome; most of the heritable forms do not respond to immunosuppressive therapy, leaving bilateral nephrectomy followed by dialysis and transplantation as the only therapeutic option.4
We report two sisters with congenital nephrotic syndrome who were born to consanguineous parents of Pakistani origin. Since no coding mutations were identified in the five aforementioned genes, we hypothesised that the girls had a novel recessive form of congenital nephrotic syndrome. Using whole exome sequencing, we discovered that both girls have an in-frame deletion in ARHGDIA that encodes the protein known as Rho GDP (guanosine diphosphate) dissociation inhibitor α (RhoGDIα, OMIM 601925).
Case history
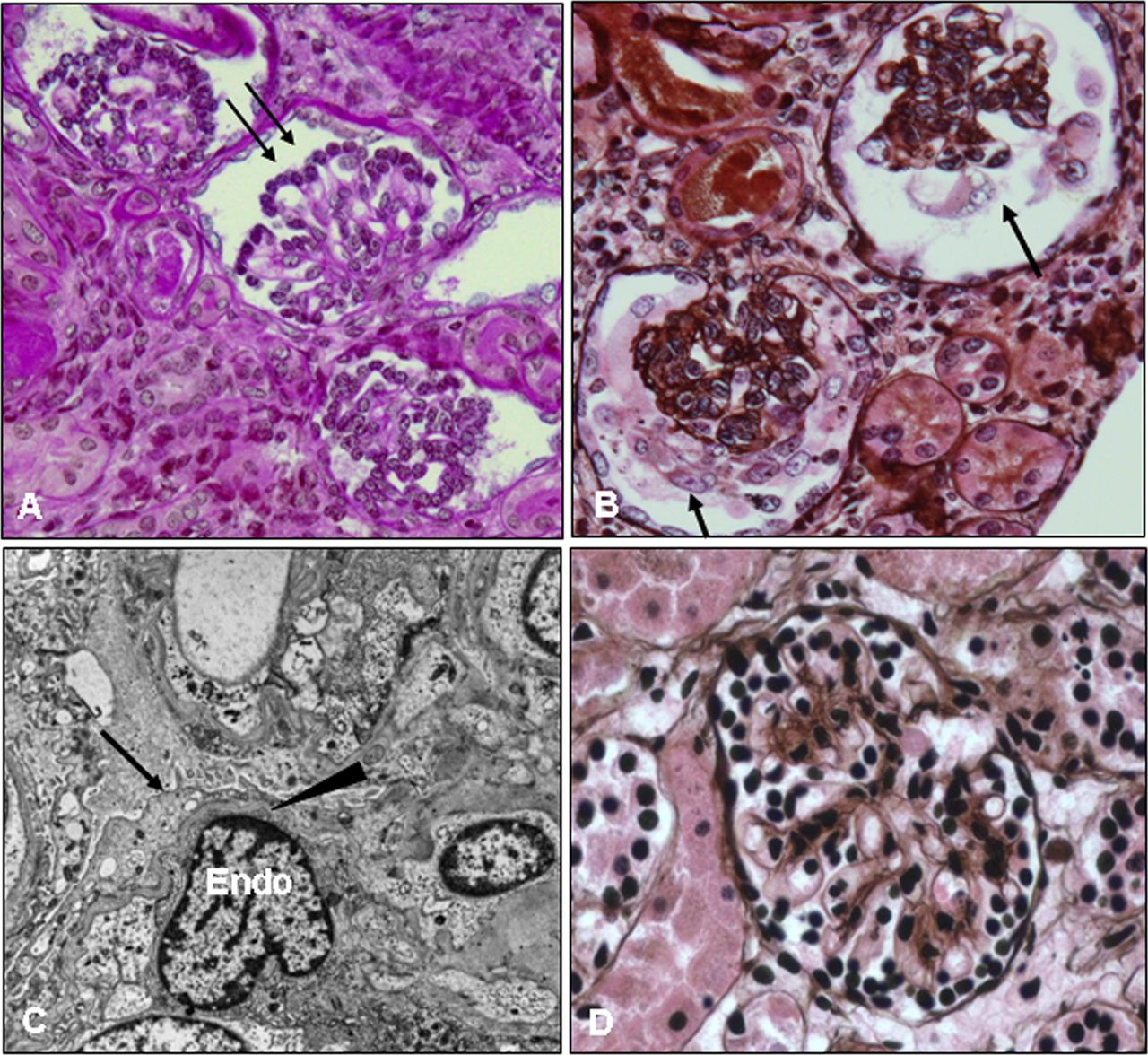
The proband presented at 3 weeks of age with generalised oedema and was found to have severe hypoalbuminaemia and proteinuria and was diagnosed with congenital nephrotic syndrome. The family history revealed that the parents were Pakistani in origin and consanguineous. A renal biopsy was performed at 1 month of age and this showed severe glomerular changes consistent with diffuse mesangial sclerosis (figure 1A–D). The patient rapidly deteriorated and developed end stage renal failure at 3 months of age that was treated by haemodialysis. She received a cadaveric renal transplant at the age of 2 years, but the graft never functioned due to venous thrombosis in the transplant, and so the patient remains on dialysis. The second child was diagnosed with congenital nephrotic syndrome on day 16 of life when her laboratory studies revealed severe hypoalbuminaemia with proteinuria (figure 2A). The mother requested no further treatment for this baby who subsequently died at the age of 2 months.
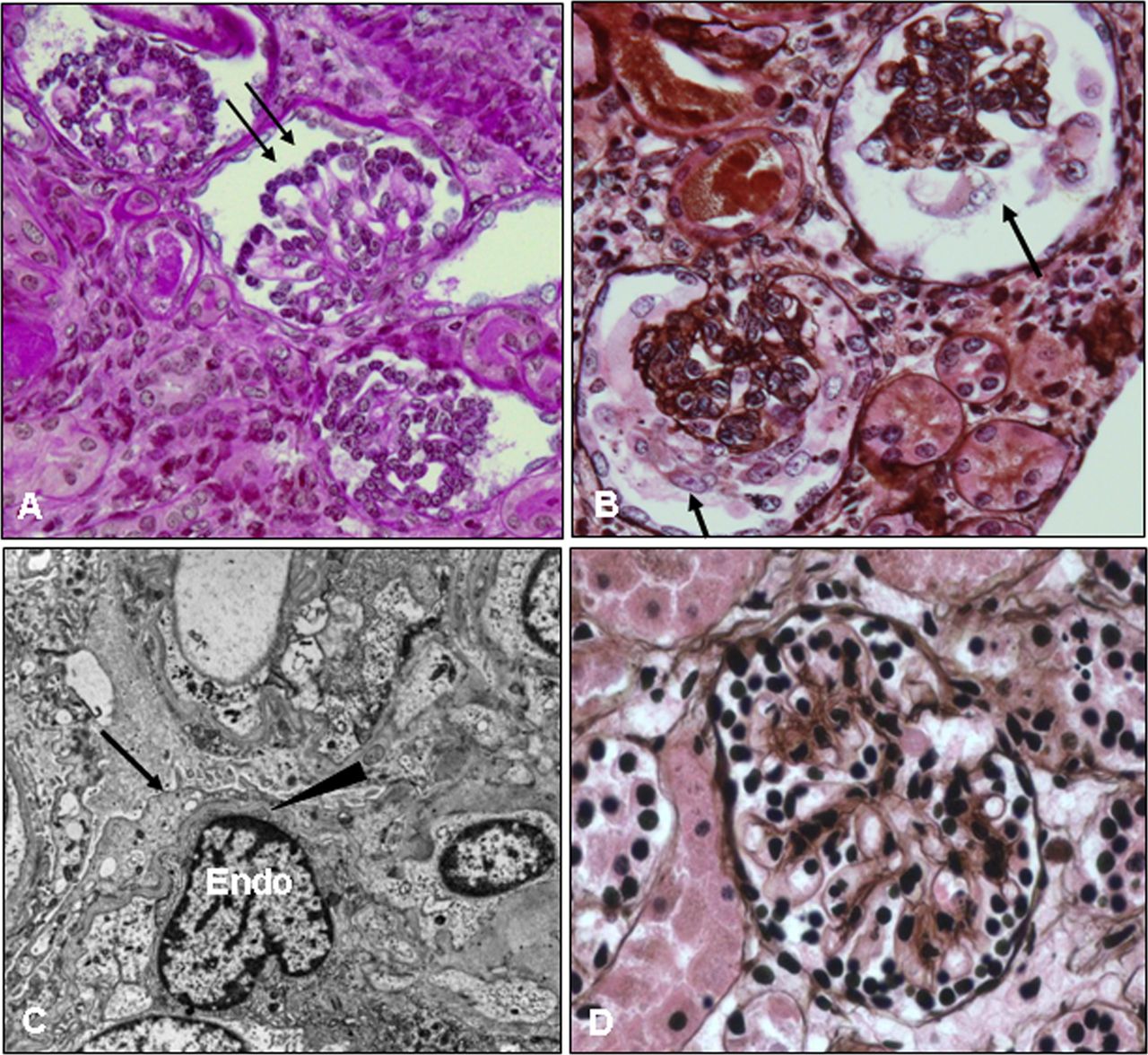
Kidney biopsy of the proband at the age of 1 month. Kidney tissue was processed and stained with periodic acid Schiff (PAS) (A) and PAS methenamine (PASM) silver (B, D) for light microscopy or for electron microscopy (C). The patient's glomeruli are notably abnormal; they are small for her age and show hypercellularity, increased extracellular matrix, and contracted/collapsed glomerular tufts that are surrounded by cuboidal/immature podocytes (A, arrows) or by vacuolated/hypertrophic podocytes (B, arrows). These changes are consistent with diffuse mesangial sclerosis. A normal glomerulus from an age matched infant is shown for comparison (D). Electron micrograph images from the patient's kidney biopsy (C) show diffuse foot process effacement (arrow), thinning of the glomerular basement membrane (arrowhead), and swollen endothelial cells (Endo). This figure is only reproduced in colour in the online version.

Family pedigree and sequencing results. (A) The parents are first cousins and have two affected daughters. (B) Whole exome sequencing results reveal the in-frame deletion in ARGHDIA in the proband and her sister, patient 2. (C) Sequencing results from the proband are shown and demonstrate that the homozygous deletion encodes one of the three consecutive aspartic acid residues (183–185) of RhoGDIα. It is not possible to determine which of the three residues is deleted and we call this mutation (c.553_555del(p.Asp185del), ΔD185). The three aspartic acid (183–185) residues are highly conserved in vertebrates. This figure is only reproduced in colour in the online version.
Methods
Ethics approval
Sequencing analyses, human fibroblast cultures and experiments were approved by the Research Ethics Board of the Montreal Children's Hospital/McGill University Health Center. Parental written informed consent was obtained for the outlined studies.
Whole exome sequencing analysis
A total of 3 μg of genomic DNA from each affected sister was used for whole exome sequencing. The DNA of patient 1, the proband, was subjected to the Agilent Sure-Select Human All Exon V.2 Kit (Santa Clara, California, USA) which targets approximately 44 Mb of the human exome defined by the National Center for Biotechnology Information (NCBI) consensus coding sequence database (CCDS, version September 2008) and additional RefSeq sequences (CCDS version September 2009). The captured exons were sequenced using two different platforms: Illumina GAIIx (San Diego, California, USA) and Illumina HiSeq2000, generating approximately 38 M (76 bp length) and 84 M (50 bp length) single-end reads, respectively. The whole exome of patient 2 was captured using the Illumina TruSeq Exome Enrichment Kit (San Diego, California, USA), which targets 62 Mb of exonic sequences including 5′UTR, 3′UTR, microRNA and other non-coding RNAs and was subsequently sequenced by Illumina HiSeq2000, producing 34 M paired-end reads with 100 bp lengths.
The bioinformatics analysis of exome sequencing data was carried out as previously described.5 ,6 Briefly, high quality trimmed reads were aligned to the human reference genome (hg19 assembly) by Burrows-Wheeler Aligner (V.0.5.9),7 producing a coverage at >5× read depth for 86% and 82% of exomes in patient 1 and patient 2, respectively.
Variants were determined using SAMtools V.0.1.7, mpileup and varFilter.8 For each called position, a minimum of two variant reads and >20% single nucleotide variants or >15% indels (small insertions or deletions) variant reads were considered. Common polymorphisms and systematic false positives were removed by filtering the variants against our in-house exome database, containing more than 500 individuals. Functional annotation of the remaining variants, those previously seen in less than five individuals in our in-house exome database, was carried out using ANNOVAR,9 which cross-references variants against public databases (dbSNP132 and the 1000 Genomes project) and annotates them according to the type of mutations (intronic, exonic, untranslated region (UTR), etc) and the score of SIFT, PolyPhen-2 and PHASTCONS tools.10 ,11 Novel variants had an allele frequency <0.5% in the 1000 Genomes dataset and were predicted to be non-synonymous (ie, missense, nonsense, frameshift, or canonical splice site changes). Candidate genes having homozygous or two potentially compound heterozygous variants were considered for further study and were manually examined using the Integrative Genomics Viewer (IGV).12
Sanger sequencing analysis
Exon 6 of ARHGDIA was amplified from gDNA in 96 ethnically matched controls using the following primers ARHGDIAE06F: AAGGCGTCAAGAGTGAGTGG and ARHGDIAE06R: GGGACACATCCGCCTGTC based on the GenBank ref seq no. NM_001185077.1.
Cell lines
A skin biopsy was performed on the proband to obtain fibroblasts for culture as described.13 Two fibroblast lines from paediatric patients without nephrotic syndrome were used as controls, MCH058 and MCH065, respectively and were obtained from the Canadian Repository for Mutant Human Cell Strains, Montreal Children's Hospital, Montreal, Quebec, Canada. All fibroblasts were maintained in α minimal essential media (αMEM) supplemented with 10% fetal bovine serum (FBS) and were serum-starved (0.5% FBS) overnight before experiments. All cell lines used were between passage 2–12. Immortalised mouse podocytes were kindly provided by Dr Shankland (University of Washington)14 and were differentiated for at least 9 days and serum-starved overnight for experiments.
Antibodies
Rabbit anti-RhoGDIα (sc-360), mouse anti-GFP (sc-9996), and rabbit anti-Myc (sc-789) were from Santa Cruz (Santa Cruz, California, USA); mouse anti-synaptopodin was from Progen Biotechnik (Heidelberg, Germany), mouse anti-RhoA, mouse anti-Rac1, and mouse anti-Cdc42 were from Upstate Biotechnologies (Lake Placid, New York, USA); rabbit anti-α-tubulin was from Abcam (Cambridge, Massachusetts, USA); Alexa Fluor 568-phalloidin was from Sigma-Aldrich (Mississauga, Ontario, Canada); 4′,6-diamidino-2-phenylindole (DAPI) was from Invitrogen (Burlington, Ontario, Canada); Alexa Fluor 488 anti-rabbit IgG was from Molecular Probes (Eugene, Oregon, USA); and Alexa Fluor 555 anti-mouse IgG was from Invitrogen (Burlington, Ontario, Canada).
Immunohistochemistry of the kidney
Kidneys from outbred adult CD1 mice were used for immunohistochemistry experiments. Kidneys were fixed in 100% methanol for 1 h at −20°C and then paraffin embedded and sectioned at 4 μm thickness. Sections were treated with citrate for antigen retrieval and stained with RhoGDIα antibody (1 : 50) and Alexa Fluor 488 anti-rabbit IgG (1 : 100). Co-staining was performed using the synaptopodin antibody (1 : 50) and Alexa Fluor 555 anti-mouse IgG (1 : 500). Photographs were taken using a Zeiss Axiophot microscope.
Plasmids
The coding sequence of human RhoGDIα was obtained by reverse transcriptase PCR (RT-PCR) using RNA from HEK293T cells and cloned into pEGFP-C1 (Clontech) using Bgl II and Hind III. The ΔD185 mutation was introduced by PCR based mutagenesis. Final sequences were confirmed by sequencing.
Immunofluorescence staining of the cells
Cells were plated on glass coverslips. For mouse podocytes, coverslips were coated with type I collagen (Sigma-Aldrich, 0.1 mg/ml, 1 h at 37°C). Cells were serum-starved overnight before the experiments, fixed in 4% paraformaldehyde, permeabilised in 0.1% Triton X-100 in phosphate buffered saline (PBS), and blocked in 3% bovine serum albumin (BSA) in PBS. Immunofluorescent detection of RhoGDIα protein was performed using RhoGDIα antibody (1 : 100) and Alexa Fluor 488 anti-rabbit IgG (1 : 500). DAPI (4’, 6-diamidino-2-phenylindole) was used at 0.06 µg/ml. Photographs were taken at 630× magnification using a Zeiss LSM 780 confocal microscope.
Rho-GTPase pull-down assay
The pull-down assay was performed as described previously using the rhotekin Rho-binding domain fused to GST (glutathione S-transferase) (GST-RBD) for RhoA and the Cdc42/Rac interactive binding domain fused to GST (GST-CRIB) for Rac1 and Cdc42.15 These fusion proteins bind specifically to the active (GTP bound), but not the inactive (GDP bound) form of Rho-GTPases, thus the assay is used to quantify the active form of Rho-GTPases.
Wound healing assay
The wound healing assay was performed under non-proliferative conditions as described previously.16 Briefly, cells were serum-starved (0.5% FBS) overnight before the experiment. Fibroblasts and mouse podocytes do not proliferate under these conditions (not shown). A scratch was created in a confluent monolayer of cells using a sterile 10 μl pipette tip. Loosely adherent cells were washed away by two vigorous washes with PBS. The cells were transferred to culture medium and photographs were taken at various time points. The percentage of wound closure was measured in four different areas, averaged, and used to quantify cell motility.
RhoGDIα knockdown
shRNAs directed to human RhoGDIα (clone ID TRCN0000106160, figure 3B(a), clone ID TRCN0000106162-4, figure 3B(b–d)) and control shRNA (Cntl) were obtained from Thermo Scientific (Rockford, Illinois, USA). Lentivirus was packaged in HEK293T cells using a standard protocol. Virus-containing medium was added to the culture medium of mouse podocytes (in permissive conditions) for 16 h and puromycin was added 48 h later. Puromycin-resistant cells were pooled and used for further experiments. The shRNA that showed the most effective knockdown (figure 4B(a)) anneals to the 3′ non-coding region of the RhoGDIα mRNA transcript.
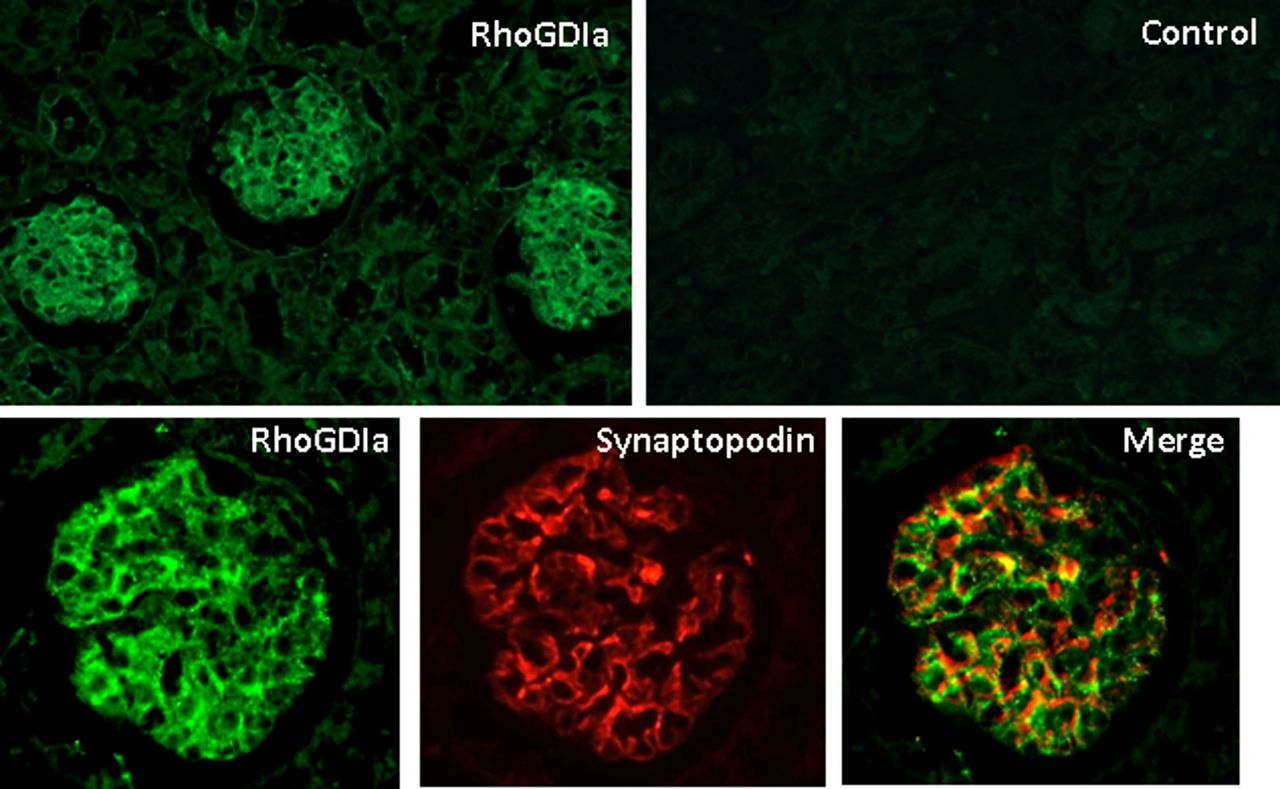
RhoGDIα is expressed in the glomerulus and in podocytes in the adult mouse kidney. Adult mouse kidney sections (methanol fixed and paraffin embedded) were stained for RhoGDIα. Sections processed with the secondary antibody alone reveal no signal (Control). Top panel, magnification 200×, bottom panel, magnification 630×. RhoGDIα is predominantly expressed in the glomerulus and in all cell types (top). However, RhoGDIα protein (green) co-localises with the podocyte protein, synaptopodin (red) as shown in the merged images (yellow signal), indicating it is expressed in podocytes. This figure is only reproduced in colour in the online version.
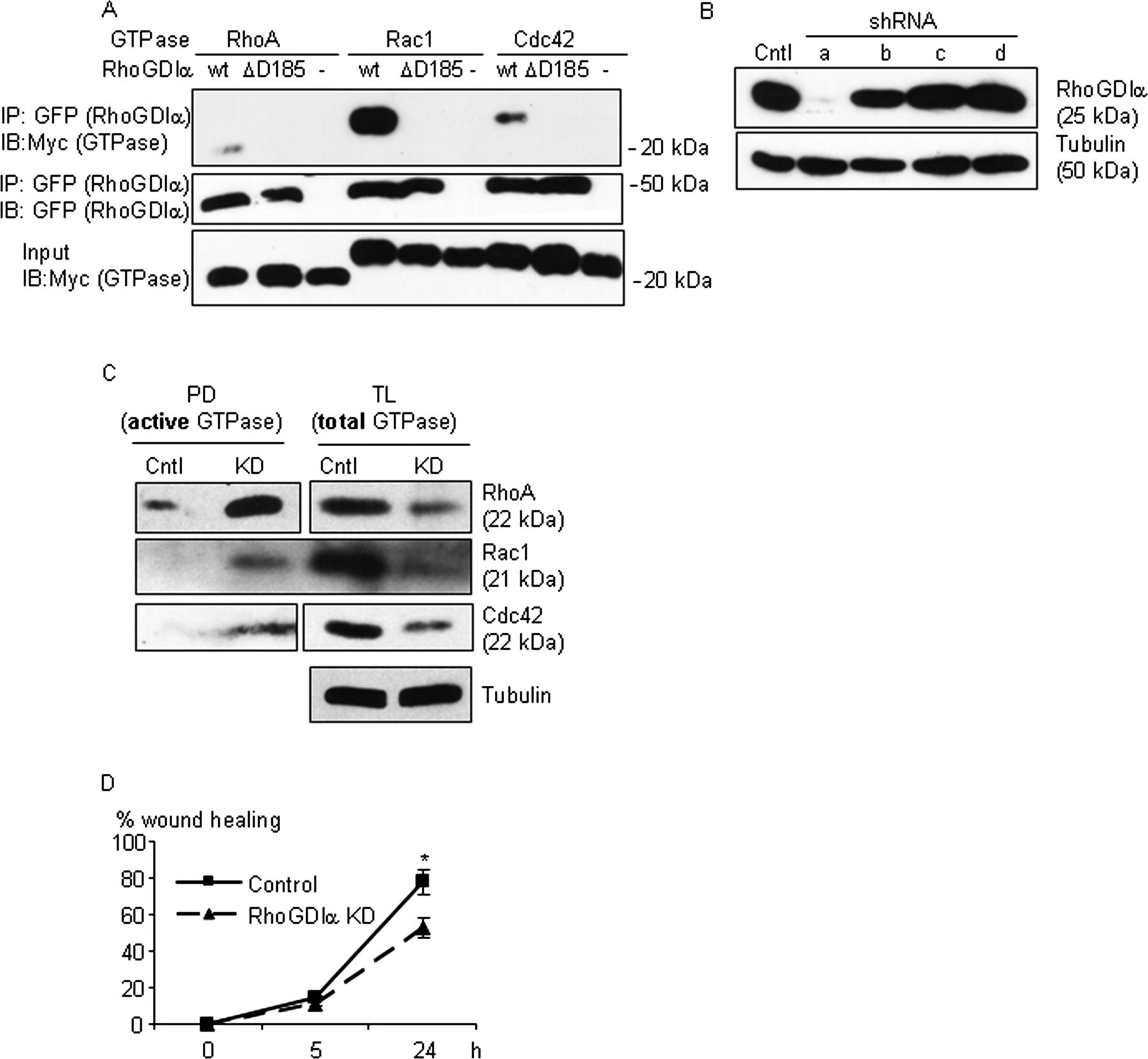
The ΔD185 protein leads to a loss-of-function and RhoGDIα knockdown (KD) leads to hyperactivation of Rho-GTPases and impaired migration of podocytes. (A) HEK293T cells were transfected with DNA constructs encoding the wild-type protein, the ΔD185 RhoGDIα proteins (GFP-tagged), or no construct (-) with Rho-GTPases, RhoA, Rac1, or Cdc42 (Myc-tagged) as indicated. Cell lysates were immunoprecipitated with anti-GFP antibody, and the precipitates and total lysates were immunoblotted for Myc (GTPases) or GFP (RhoGDIα). Wild-type RhoGDIα, but not the ΔD185 protein, co-immunoprecipitated with the three GTPases (representative of three experiments). (B) Immortalised mouse podocytes were transduced with four RhoGDIα shRNAs or control shRNA. Cells were selected with puromycin and pooled, lysed, and immunoblotted for RhoGDIα. One of the four shRNAs tested (a) showed effective KD. (C) Levels of active RhoA, Rac1, and Cdc42 detected by pull-down assay (PD, Methods) were increased, whereas the amounts of total Rho-GTPase proteins were decreased in KD cells. Representative blots of at least three experiments for each GTPase are shown. (D) The wound healing assay showed impaired cell motility of the KD cells. *p<0.02 vs KD, n=4.
Statistics
Data are presented as mean±SEM. The t statistic was used to determine significant differences between two groups. One-way analysis of variance was used to determine significant differences among groups. When significant differences were found, individual comparisons were made between groups using the t statistic and by adjusting the significance threshold according to the Bonferroni method.
Results
Mutation in ARHGDIA leads to congenital nephrotic syndrome
Whole exome sequencing was performed and after filtering out common and non-protein coding variants, 438 rare variants were identified in the proband (see Methods) (figure 2B). Based on parental consanguinity, the mode of inheritance was assumed to be autosomal recessive, thus only candidate genes having homozygous or two or more potentially compound heterozygous variants were considered for further study. Due to the poor quality of the archived DNA, we were only able to perform partial whole exome sequencing in patient 2 such that the exome coverage was 10×. However, this result in combination with manual visualisation of the data was sufficient to exclude most of the candidates, and reduce the list to five genes. Further targeted Sanger sequencing of regions that could not be resolved in the low coverage exome sequencing excluded three additional variants and resulted in a list of two candidate genes: one homozygous variant, ARHGDIA, and one compound heterozygous variant, TSGA10, shared between the proband and her sister. We did not pursue TSGA10 since one of the alleles (c.G568A) was not predicted to be damaging by PolyPhen2 and SIFT algorithms, and the gene did not appear to explain the renal phenotype. Several lines of evidence clearly pointed to ARHGDIA as the gene responsible for the observed phenotype. Mice lacking ARHGDIA develop massive proteinuria and nephrotic syndrome and die from renal failure as adults.17 ,18 Furthermore, mutations in ARHGDIA were recently identified in two unrelated children with glucocorticoid-resistant nephrotic syndrome.19
Both sisters harbour a homozygous in-frame deletion of a single amino acid at chr17 : 79826812-79826814 genomic position, corresponding to c.553_555del (p.Asp185del) by HGVS (Human Genome Variation Society) annotation (figure 2B). The in-frame deletion has not been previously detected in our exome database (>500 exomes), dbSNP, 1000 Genome Project or the EVS (Exome Variant Server) databases, and was subsequently confirmed by Sanger sequencing in both sisters (figure 2C). The deletion is predicted to remove one of three consecutive highly conserved aspartic acid residues, D183, 184, or 185 in the protein, Rho GDP dissociation inhibitor α (RhoGDIα). The girls’ mother is healthy and was found to be a heterozygous carrier for this deletion. The father's DNA was unavailable for analysis. The deletion was not found in 96 ethnically matched subjects that were sequenced (data not shown).
RhoGDIα is ubiquitously expressed with high levels in the lung, the thymus, the spleen, the small intestine, and the kidney.20 It regulates small GTP binding proteins of the Rho/Rac/Cdc42 family which are critical for the function of the actin cytoskeleton.21 ,22 We therefore embarked on further studies to establish that the c.553_555del in ARHGDIA, referred to as ΔD185 in the subsequent functional studies, was responsible for congenital nephrotic syndrome in the two sisters.
RhoGDIα is expressed in podocytes in the adult kidney
To determine if RhoGDIα protein was expressed within podocytes, a highly specialised cell that is critical for the function of the glomerular filtration barrier,23 immunohistochemistry was performed. RhoGDIα protein was strongly expressed in the glomerulus of the adult mouse kidney, where it co-localised with synaptopodin, a specific marker of podocytes24 (figure 3). RhoGDIα protein was also detected in other cell types within the glomerulus, including mesangial cells, as reported previously.25
RhoGDIα ΔD185 leads to loss-of-function
Structural analysis of the RhoGDIα protein indicates that D184 and D185 are located at the interface where RhoGDIα interacts with Rho-GTPases, suggesting that the ΔD185 protein would generate a loss-of-function phenotype.26 DNA constructs encoding the wild-type and the ΔD185 proteins were transiently transfected in HEK293T cells. The wild-type RhoGDIα protein co-immunoprecipitated with the three Rho-GTPases, RhoA, Rac1, and Cdc42, while the mutant failed to do so, supporting the hypothesis that the ΔD185 deletion results in a loss-of- function (figure 4A).
Loss-of-function of RhoGDIα causes hyperactivation of Rho-GTPases and impairs podocyte motility
To determine the effect of the loss-of-function of RhoGDIα in podocytes, we knocked down the endogenous protein in cultured mouse podocytes using shRNA (figure 4B). Podocyte cells with knockdown of RhoGDIα protein demonstrated much higher levels of activated RhoA, Rac1, and Cdc42 as compared to control cells, demonstrating that in the absence of functional RhoGDIα, Rho-GTPases were no longer maintained in their inactive state (figure 4C). A global decrease in the amount of total RhoA, Rac1, and Cdc42 was also noted with the knockdown of RhoGDIα (figure 4C), consistent with a recent report demonstrating that Rho-GTPases undergo rapid proteosomal degradation in the absence of RhoGDIs.27 Altered podocyte cell motility has been shown to correlate with the development of proteinuria.28–30 To determine if hyperactivation of RhoA, Rac1, and Cdc42 affected cell motility, wound healing assays were performed. Podocyte cells with knockdown of RhoGDIα demonstrated impaired wound healing when compared with control cells (figure 4D). In summary, a loss-of-function of RhoGDIα results in hyperactivation of RhoA, Rac1, and Cdc42 and leads to impaired podocyte motility, likely via altered actin dynamics.
RhoGDIα ΔD185 leads to hyperactivation of Rho-GTPases and impairs fibroblast cell motility
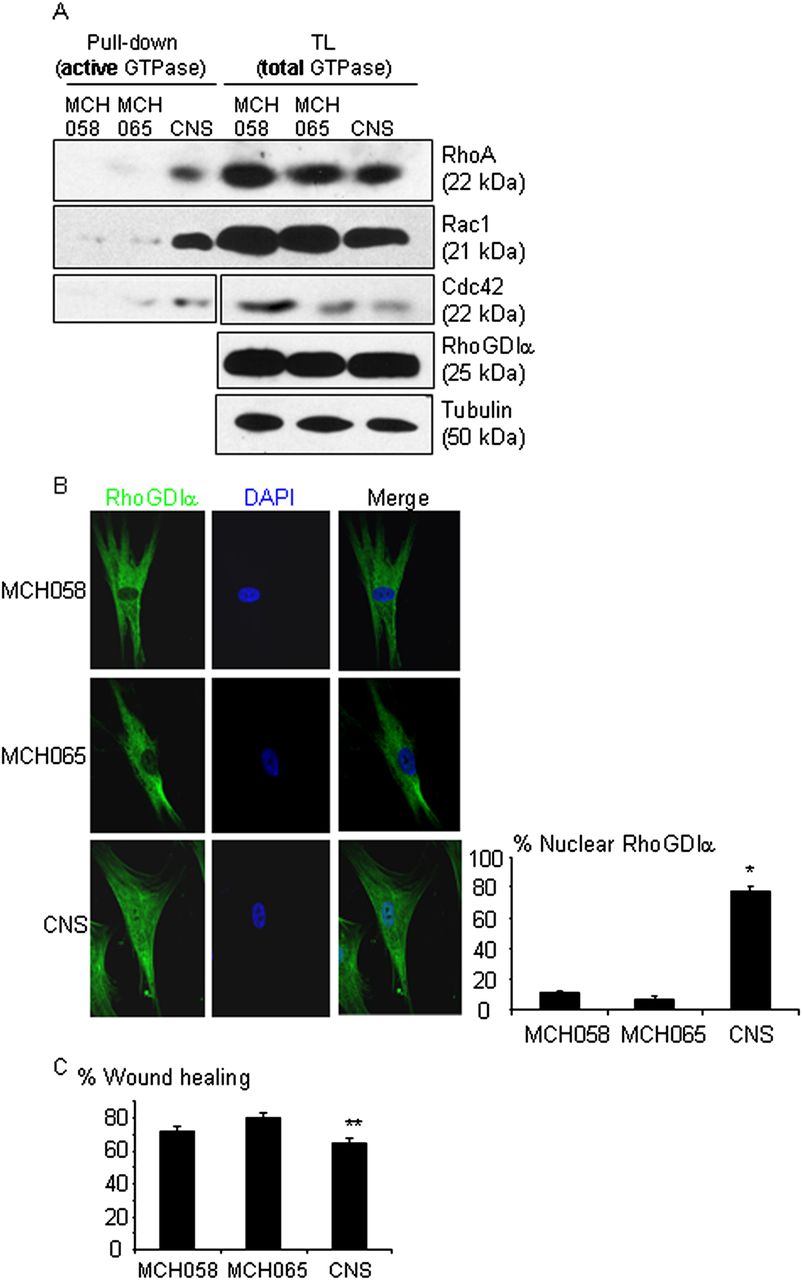
To evaluate the function of the endogenous wild-type and ΔD185 proteins, we obtained skin fibroblasts from the proband. Fibroblast cell lines from a healthy boy (MCH058) and girl (MCH065) were used as controls. In the proband's fibroblasts, RhoGDIα protein was expressed at a similar level as compared with control cells (figure 5A). Levels of activated RhoA, Rac1, and Cdc42 were notably higher in the proband's fibroblasts as shown by pull-down assay, when compared with control cells (figure 5A), indicating hyperactivation of all three Rho-GTPases, due to the lack of function of the ΔD185 protein. Immunofluorescent detection of the RhoGDIα protein revealed that it was distributed predominantly in the cytosol in control fibroblasts, consistent with previous reports,21 ,22 whereas in the proband's fibroblasts, the protein was also detected in the nucleus, indicating that the ΔD185 protein was mislocalised (figure 5B and online supplementary figure S1). Similar nuclear mislocalisation was observed when ΔD185 was overexpressed in podocytes (data not shown). Cell motility was quantified using the wound healing assay. Similar to what was observed in podocytes with RhoGDIα knockdown, the proband's fibroblasts migrated more slowly, as compared with control cells (figure 5C). Taken together, the in-frame deletion in ARHGDIA observed in the two patients leads to a loss-of-function phenotype characterised by hyperactivation of Rho-GTPases and impaired cell motility.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The ΔD185 protein leads to a loss-of-function, hyperactivation of Rho-GTPases and impaired migration in fibroblasts from the proband. Control human skin fibroblasts (MCH058 and MCH065) and the proband's skin fibroblasts (CNS) were compared. (A) RhoA, Rac1 and Cdc42 activities detected by pull-down assay were notably increased in the proband's fibroblasts, compared with control cells. (B) Immunostaining of RhoGDIα showed a predominantly cytosolic distribution in control cells and nuclear mislocalisation in the proband's fibroblasts. The graph shows the percentage of cells with nuclear staining of RhoGDIα. Approximately 40 cells were counted in each experiment for each cell line. n=4 experiments, *p<0.01 versus two control cells. (C) The wound healing assay showed impaired cell motility of the proband's fibroblasts. The graph is the average of four independent experiments. **p<0.01 vs MCH065 and p=0.06 vs MCH058. This figure is only reproduced in colour in the online version.
Discussion
The current study has identified ARHGDIA as a new gene implicated in the pathogenesis of autosomal recessive congenital nephrotic syndrome. The mutation leads to a loss-of-function of RhoGDIα, which results in the hyperactivation of Rho-GTPases and impaired cell motility as shown in the proband's fibroblasts and in podocytes when RhoGDIα was knocked-down. Our findings are consistent with the phenotype of RhoGDIα knockout mice that develop heavy proteinuria and progressive kidney failure, leading to death at 1 year of age.18 Histologically, the glomeruli of these mice show a variable degree of glomerular sclerosis with obliteration of the capillary loops, accumulation of extracellular matrix and podocyte injury manifested as foot process effacement.18 All of these histological features were also observed in the proband's renal biopsy (figure 1). The mouse model, in combination with our findings in the two patients, demonstrates that RhoGDIα is critical for the function of the glomerular filtration barrier. We propose that loss-of-function of RhoGDIα disturbs the balance of Rho-GTPases within podocytes and this causes derangement of the actin cytoskeleton, podocyte injury, and nephrotic syndrome.
The history of parental consanguinity and the absence of disease in either parent suggests that the disease is being transmitted in an autosomal recessive inheritance pattern. While we cannot completely rule out the possibility that there are parent-of-origin effects like imprinting or sex-limited expression or that the disease is being transmitted via the X chromosome with X inactivation, the recent discovery by Gee et al19 of two unrelated children with homozygous mutations in ARHGDIA and nephrotic syndrome leads us to believe that the recessive transmission of c.553_555del in ARHGDIA is the causal gene defect in the two sisters.
The link between Rho-GTPase dysregulation, podocyte injury and nephrotic syndrome is well established. In mice, inducible expression in podocytes of a constitutively active or a dominant negative RhoA both induced proteinuria,31 ,32 while podocyte specific deletion of Cdc42 caused congenital nephrotic syndrome.33 The urokinase receptor (suPAR), whose soluble form is a potential candidate for the permeability factor believed to cause idiopathic focal segmental sclerosis (FSGS), induces actin cytoskeletal derangement via Rac1 and Cdc42 activation.34 ,35 Rac1 activation in podocytes has been implicated in the pathogenesis of HIV associated nephropathy,36 and a loss-of-function mutation of ARHGAP24 that encodes a Rac-1 GTPase activating protein was recently reported to cause adult onset FSGS, likely via hyperactivation of Rac1 in podocytes.28 It is therefore plausible that Rho-GTPases contribute not only to the pathogenesis of hereditary forms of nephrotic syndrome, but also to other forms including idiopathic minimal change disease and FSGS. Further studies that examine individual Rho-GTPases will be important pursuits to elucidate the relationship between the actin cytoskeleton and podocyte function in these disorders.
Approximately 15% of children with congenital nephrotic syndrome fail to demonstrate protein altering variants in NPHS1, NPHS2, WT1, and LAMB2, suggesting that mutations in additional genes need to be considered, such as ARHGDIA.1 Although our patients presented with congenital nephrotic syndrome associated with a critical deletion in the interface region where RhoGDIα binds to Rho-GTPases, it is possible that other mutations in this gene might present with later onset forms of nephrotic syndrome as has been reported for NPHS1, NPHS2, and LAMB2.37–39
In summary, we have identified ARHGDIA as a new gene implicated in congenital nephrotic syndrome. Mutations in ARHGDIA are predicted to cause an imbalance in the active and inactive forms of Rho-GTPases leading to derangements in the actin cytoskeleton within podocytes and nephrotic syndrome that may present in the first year of life or later, depending on the biological impact of the mutation. Thus, we believe that genetic screening for ARHGDIA mutations should be considered in children with nephrotic syndrome when there is a family history suggesting recessive inheritance.
Acknowledgments
We thank the family for agreeing to this study. This work was supported by grants from the Canadian Institute of Health Research (MOP-53335 to TT) and the Montreal Children's Hospital Research Institute (to IRG and MB). We acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Genome Québec Innovation Centre (Montréal, Canada). Exome sequencing for this work was supported by RaDiCAL.40 JM is a recipient of a Canada Research Chair. IRG and TT hold scholarship awards from the Fonds de la Recherche en Santé du Québec. The authors thank all of the paediatric nephrologists, the geneticist Dr L Russell, the genetic counsellor Ms R Vanneste, and all nursing staff, especially Ms S Champoux, who were involved in the care of these patients.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure 1
Footnotes
CB, DA, KCH, JEA and SF contributed equally.
-
Contributors IRG oversaw the study, was involved in conceptual design, drafting and editing the manuscript and is a guarantor of the work; CB generated the data and created the following figures: figure 4–C, and figure 5A; DA generated the data and created the following figures: figure 4D, figure 5B, 5C, and supplementary figure S1; KH analysed the original WES data on the proband and identified a candidate list of homozygous variants; JEA cultured the patient's fibroblasts and generated the data used to create figure 3; SF compared the WES data for the two sisters, analysed the Sanger sequencing performed on the proband, the sister and the mother, and created figure 2; MB recruited the proband, initiated the original genetic analysis for candidate genes, and was involved in manuscript editing; CB analysed the patient's kidney biopsy and created figure 1; MRA performed the sequencing of 96 ethnically matched controls for ARHGDIA; SAN oversaw the sequencing and was responsible for recruiting this cohort; DSR was involved in editing the manuscript; JM oversaw the WES analysis, the sequencing, and was involved in conceptual design and in editing the manuscript; TT oversaw the study, was involved in conceptual design, wrote the first draft of the manuscript, and was involved in editing the manuscript and is a guarantor of the work.
-
Competing interests None.
-
Ethics approval Research Ethics Board of the Montreal Children's Hospital/McGill University Health Center.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/