Article Text

Abstract
Background Isolated complex II deficiency is a rare form of mitochondrial disease, accounting for approximately 2% of all respiratory chain deficiency diagnoses. The succinate dehydrogenase (SDH) genes (SDHA, SDHB, SDHC and SDHD) are autosomally-encoded and transcribe the conjugated heterotetramers of complex II via the action of two known assembly factors (SDHAF1 and SDHAF2). Only a handful of reports describe inherited SDH gene defects as a cause of paediatric mitochondrial disease, involving either SDHA (Leigh syndrome, cardiomyopathy) or SDHAF1 (infantile leukoencephalopathy). However, all four SDH genes, together with SDHAF2, have known tumour suppressor functions, with numerous germline and somatic mutations reported in association with hereditary cancer syndromes, including paraganglioma and pheochromocytoma.
Methods and results Here, we report the clinical and molecular investigations of two patients with histochemical and biochemical evidence of a severe, isolated complex II deficiency due to novel SDH gene mutations; the first patient presented with cardiomyopathy and leukodystrophy due to compound heterozygous p.Thr508Ile and p.Ser509Leu SDHA mutations, while the second patient presented with hypotonia and leukodystrophy with elevated brain succinate demonstrated by MR spectroscopy due to a novel, homozygous p.Asp48Val SDHB mutation. Western blotting and BN-PAGE studies confirmed decreased steady-state levels of the relevant SDH subunits and impairment of complex II assembly. Evidence from yeast complementation studies provided additional support for pathogenicity of the SDHB mutation.
Conclusions Our report represents the first example of SDHB mutation as a cause of inherited mitochondrial respiratory chain disease and extends the SDHA mutation spectrum in patients with isolated complex II deficiency.
- Neurology
- Mitochondrial disease
- Genetic screening/counselling
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
In eukaryotic cells, the mitochondrial oxidative phosphorylation (OXPHOS) pathway is the primary mechanism for ATP production. This OXPHOS system comprises five transmembrane complexes consisting of ∼90 protein subunits that are encoded by the mitochondria's own genetic material (mtDNA) and the nuclear genome. Electrons, supplied by the oxidation of fat and carbohydrates, are transferred through the mitochondrial respiratory chain complexes to complex V, where oxygen acts as the ultimate electron acceptor. Coincident with electron transfer is the extrusion of protons across the inner membrane from the matrix to the intermembrane space, creating an electrochemical gradient which is dissipated through complex V (ATP synthase) driving the phosphorylation of ADP to form ATP. Mitochondrial respiratory chain disease represents a major inborn error of metabolism and is caused by defective OXPHOS.1 Mitochondrial disease is associated with both a varied age of onset and a diverse phenotypic spectrum.2 The hallmark clinical and genetic heterogeneity is often compounded further by the lack of clear genotype–phenotype correlations. Biochemical assessment of respiratory chain complex activities can often provide guidance for the genetic testing strategy, but uncovering the underlying genetic defect can be difficult. For many patients, especially children, the genetic aetiology of their condition remains unknown.
Complex II differs from the other complexes of the mitochondrial respiratory chain in that its four structural subunits (SDHA, SDHB, SDHC and SDHD) and two known assembly factor genes (SDHAF1 and SDHAF2) are all nuclear-encoded. The soluble flavoprotein (Fp/SDHA) and iron–sulphur (Fe-S/SDHB) proteins, encoded by SDHA and SDHB, respectively, have catalytic activity and together form succinate dehydrogenase (SDH), while the SDHC and SDHD subunits act to anchor the complex to the inner mitochondrial membrane and its interaction with the quinone pool.3 Complex II is also unique in that it is part of both the respiratory chain and the Krebs cycle.
Mitochondrial disease presentations associated with an isolated deficiency of complex II are rare, accounting for an estimated 2% of respiratory chain deficiencies.4 ,5 Reported cases have presented in childhood with Leigh syndrome,4 ,6–8 a fatal respiratory disease with severe hypoglycaemia,9 neonatal cardiomyopathy10 and an infantile leukoencephalopathy.11 The only reported exception to these childhood presentations is the report of two sisters with an adult-onset phenotype characterised by progressive optic atrophy, ataxia and myopathy.12
In addition to primary mitochondrial disease presentations, germline mutations in SDHA,13 SDHB,14 SDHC,15 SDHD16 ,17 and SDHAF218 are recognised causes of familial pheochromocytomas and paragangliomas, thus establishing a link between SDH deficiency and susceptibility to tumourigenesis.19 The factors determining whether SDH defects lead to neurological disease or impaired tumour suppression are poorly understood, yet both are related to loss of enzyme activity and perturbation of the complex formation. On account of its similarity with the human enzyme, the Saccharomyces cerevisiae has proven a useful model system to study the effects of SDH gene mutations, in particular germline missense mutations associated with paraganglioma development.20
Here, we report two paediatric patients presenting with leukoencephalopathy with isolated complex II deficiency in whom molecular investigations revealed novel compound heterozygous p.Thr508Ile and p.Ser509Leu SDHA mutations in one patient, and a novel, homozygous p.Asp48Val SDHB mutation in the second. This represents the first example of SDHB-related pathology in association with a primary mitochondrial disease presentation, with pathogenicity confirmed by functional modelling studies in yeast.
Subject and methods
Patient 1
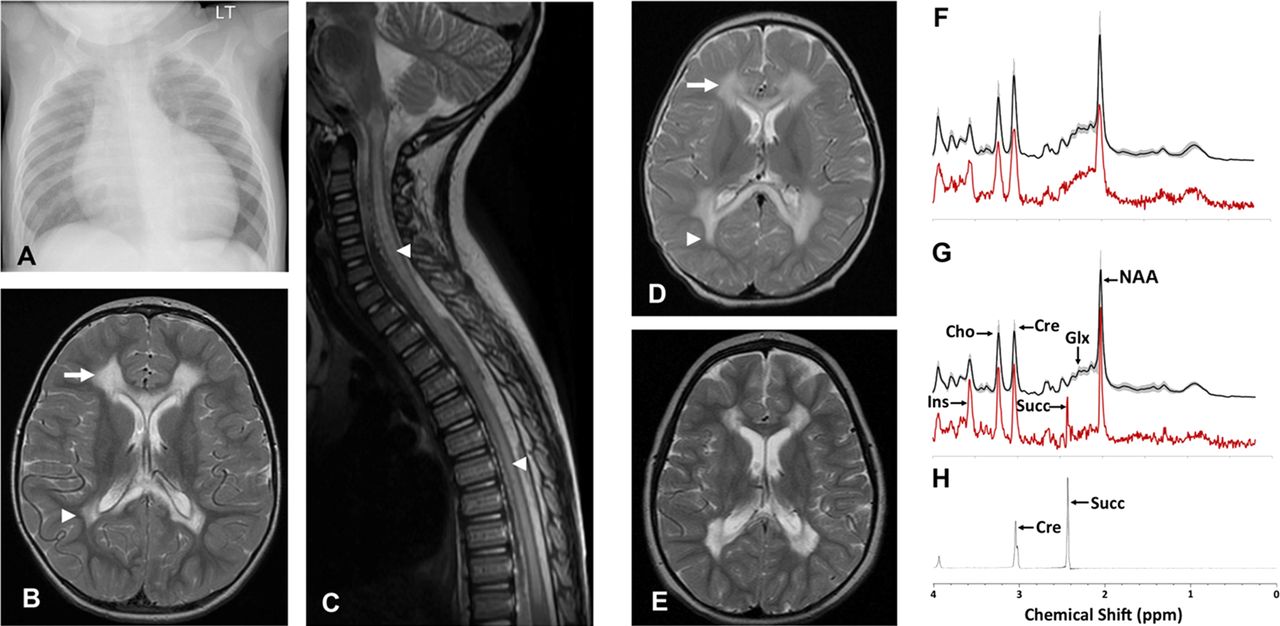
Patient 1 is a male child born at term to non-consanguineous, mixed ethnicity parents following an uncomplicated pregnancy. He was well during the neonatal period, but at 3 months of age he presented with increasing dyspnoea, sweating and difficulty feeding. Cardiomegaly was evident on chest radiograph (figure 1A) while echocardiogram demonstrated ischaemia and a dilated and markedly hypertrophied left ventricle, with a degree of non-compaction of the left ventricular myocardium and a fractional shortening of only 10%. Cardiac catheter investigation demonstrated no abnormality of the coronary arteries, and he was diagnosed with dilated cardiomyopathy. Cardiac function improved and stabilised with short-term inotropic support, ongoing diuretic therapy and ACE inhibitors.

(B) Axial T2-weighted MR image of Patient 1’s brain demonstrates high signal intensity in frontal (arrow) and peritrigonal (arrow head) white matter sparing subcortical U-fibres, normal basal ganglia whilst the sagittal T2-weighted spinal image (C) demonstrates extensive high signal intensity in spinal cord grey matter (arrow heads).
Initial development was satisfactory so that by 8 months of age he could sit unsupported, roll over, pull to stand and was babbling. However, motor delay was noted by 18 months of age. At review at 2.5 years of age, he was able to stand, but with knees and hips in flexion due to contractures. He had a few single words of speech. Examination revealed generalised hypertonia in all limbs with brisk deep tendon reflexes and bilateral upgoing plantar response.
Cranial and spinal MRI at 2.5 years age (figure 1B) demonstrated extensive cystic change and abnormal high T2 and FLAIR signal in the central cerebral white matter symmetrically and bilaterally, with scattered peripheral foci of abnormal signal in the frontal lobes. There was also abnormal signal in the anterior and posterior corpus callosum, ventral pons, medulla and throughout the majority of the grey matter of the spinal cord (figure 1C). Metabolic investigations included normal random plasma lactate (1.5 mmol/l). Acylcarnitine profile demonstrated increased acetyl and hydroxybutyryl carnitines consistent with ketosis/lactic acidosis, with a normal free carnitine concentration. Urinary amino acids were normal, while organic acids demonstrated raised lactate, ketones and 3-hydroxyisovalerate and elevated tricarboxylic acid cycle metabolites (succinate, fumarate and 2-ketoglutarate). A diagnostic open skeletal muscle biopsy was obtained at 3 years of age.
Patient 2
Patient 2 is a female child born at term to consanguineous Asian parents. There were no antenatal or perinatal complications, no previous history of miscarriages, and two older siblings are alive and well. Growth and developmental milestones were normal until 1 year of age by which time she could cruise around furniture and was babbling. Subsequently, over a 6-week period, she lost the ability to walk, became unsteady and had repeated falls. There was no associated head injury or febrile illness. She became very hypotonic with poor head control and difficulty feeding. After the 6-week period of deterioration she made slow developmental progress, and there were no further episodes of developmental regression. By the age of 4 years, she could stand briefly with support although remains wheelchair-dependent, having developed flexion contractures in both her arms and legs.
MRI acquired at the initial presentation (figure 1D) revealed leukodystrophy with extensive signal changes in the deep cerebral white matter, sparing U-fibres, with abnormalities also present in cerebellum and brainstem. A repeat brain MRI study at 4.5 years (figure 1E) revealed a similar pattern of abnormalities but with cyst formation in the abnormal white matter, and persistent signal abnormalities in the corpus callosum. The short echo time MR spectroscopy of the dystrophic white matter (figure 1G) demonstrated the presence of a singlet lactate peak at 2.4 ppm, seen also in long echo time (not shown) but not detected in a control group or in the normal appearing basal ganglia of patient 2 (figure 1H),21 confirming the accumulation of succinate in the dystrophic white matter in vivo. The metabolite profile of the white matter also demonstrated significantly decreased glutamine and glutamate, relative preservation of the neuronal marker N-acetylaspartate and increased myo-inositol compared with a previously reported age-matched cohort.22 The metabolite profile of the unaffected basal ganglia appeared relatively normal with no succinate detected (figure 1F).
Metabolic investigations demonstrated normal plasma, very long chain fatty acids, carnitine profile, amino acids, ammonia and lactate. Urinary organic and amino acids, glycosaminoglycans and oligosaccharides were also normal, as were cerebrospinal fluid glucose and lactate. A diagnostic open skeletal muscle biopsy was obtained at age 5 years.
Muscle histology and biochemistry
Informed consents with appropriate ethics review committee approvals were obtained. Histological and histochemical analyses were performed on 10 µm transversely-orientated serial cryosections of skeletal muscle biopsy samples using standard procedures. The activities of individual respiratory chain complex activities and citrate synthase, a mitochondrial matrix marker, were determined in muscle homogenates as previously described.23
Molecular genetics
Total genomic DNA was obtained using standard methods and the coding region plus intron–exon boundaries of the SDHA, SDHB and SDHAF1 genes were amplified using locus specific primers (sequences available upon request). Amplicons were sequenced using the BigDye v3.1 kit and capillary electrophoresed on the ABI3130×l fluorescent sequencing platform (Life Technologies, Warrington, UK). Chromatograms were compared with appropriate GenBank reference sequences (SDHAF1: NM_001042631.2; SDHA: NM_004168.2; and SDHB: NM_003000.2). All sequence variants were cross-referenced against dbSNP (build 135) and all variants of unknown pathological significance were investigated using in silico methodologies.24,27
In silico prediction tools
Amino acid residue conservation and predicted impact of the novel SDHA and SDHB variants were investigated using Ensembl release 66,24 Polyphen2,25 SIFT26 and AlignGVGD.27 Putative effects of the novel SDHB variant on SDHB tertiary structure were proposed using Phyre2,28 while residue interactions between the SDH subunits were characterised using Piccolo.29 Sequence alignment for mutation analysis was performed with Clustal30 and BLAST.31
BN-PAGE and SDS-PAGE
Blue native polyacrylamide gel electrophoresis (BN-PAGE) was used to investigate the native structures of respiratory chain enzymes. For BN-PAGE, the NativePAGE Novex Bis-Tris Gel and blot transfer system was used and samples were run using precast 4%–16% Bis-Tris gels (Invitrogen, Carlsbad, California, USA). For Patient 2 and two aged-matched controls, enriched mitochondria were prepared from muscle using differential centrifugation after homogenisation in Medium A (120 mM KCl, 20 mM HEPES, 5 mM MgCl2, 1 mM EGTA, pH 7.2). For Patient 1 and two separate paediatric controls, mitochondria were isolated from cultured fibroblasts as described32 using anti-TOM22 coated MicroBead system (Miltenyi Biotec, Bergisch Gladbach, Germany). Protein content was determined using Bradford reagent (Bio-Rad, Hercules, California, USA) and between 2 and 10 µg of mitochondria were loaded, depending on the postrun analysis. For Patient 1 and controls, complex I ingel activity analysis was performed.33 Following western blot transfer of BN-PAGE gels, complexes I and II were probed with mouse antihuman immunoglobulin directed at NDUFA9 and the flavoprotein and iron–sulphur subunits of SDH, respectively. All primary antibodies, except TOM20 (Santa Cruz, Biotechnology, Santa Cruz, California, USA), were purchased from Mitosciences/Abcam (Cambridge, UK).
Proteins were separated by SDS-PAGE, transferred and membranes probed with antibodies against SDHA, SDHB and NDUFB8 as well as porin or TOM20 (as mitochondrial loading markers). For detection, blots were treated with appropriate HRP-conjugated immunoglobulins (Dako, Glostrup, Denmark), followed by ChemiLucent detection reagents (GE Healthcare, Buckinghamshire, UK).
Yeast strains and culture conditions
Yeast strains, BY4741 (MATa; his3D1 leu2D0 lys2D0 ura3D0) and its isogenic sdh2:kanMX4 mutant, were transformed using the lithium acetate method.34 Restriction-enzyme digestions, Escherichia coli transformation and plasmid extractions were performed using standard methods.35
Cells were cultured in yeast nitrogen base (YNB) medium (0.67% YNB without amino acids (ForMedium, Hunstanton, UK)) supplemented with 1 g/l of drop-out powder36 containing all amino acids except those required for plasmid maintenance. Various carbon sources were added at 2% (w/v) (Carlo Erba Reagents, Milan, Italy). Media were solidified with 20 g/l agar (ForMedium). For respiration and mitochondria extraction, cells were grown to late-log phase in the YNB medium supplemented with 0.6% glucose.
Construction of yeast mutant alleles
The sdh2 p.Asn42Asp and p.Asn42Val mutant alleles were obtained by site-directed mutagenesis using the overlap extension technique.37 In the first set of PCR reactions, the SDH2 region was obtained using the forward primer HSDH2F-CGCGAAGCTTGCTGAGGTGCAAATGGCCACCC and the following reverse mutagenic primers RS242ND-GGTTTAGCACTTGGCTCGTCTGGGTCCCATCTGTAAACTTTAAAAG and RS242NV-GGTTTAGCACTTGGCTCGTCTGGCACCCATCTGTAAACTTTAAAAG where base changes are indicated in bold. The second SDH2 region was obtained using the forward mutagenic primers FS242ND and FS242NV, complementary to RS242ND and RS242NV, and the reverse primer SSDH2R-CCCCGTCGCCACCTTGTCGCCTATGATGG. The final mutagenised products were obtained using the overlapping PCR fragments as template with HSDH2F and SSDH2R as external primers. The products were then digested with HindIII and SalI and cloned in HindIII-SalI digested pFL38 centromeric plasmid.38
Isolation of mitochondria, enzyme assays and respiration
Oxygen uptake was measured at 30°C using a Clark-type oxygen electrode in a 1 ml stirred chamber containing 1 ml of air-saturated respiration buffer (0.1 M phthalate–KOH, pH 5.0) and 10 mM glucose (Oxygraph System, Hansatech Instruments, England). The reaction was initiated with the addition of 20 mg of wet weight of cells, as described.20 Preparation of mitochondria and SDH activity (expressed as nmol/min/mg protein) were also performed as described.20
Results
Histological, histochemical and respiratory chain analyses
The histological examination of diagnostic skeletal muscle biopsies was normal for both patients, with the exception of a few atrophic fibres in Patient 1, and a subtle increase in intrafibre lipid content for Patient 2 (figure 2). Histochemical analysis of both patients’ biopsies demonstrated normal cytochrome c oxidase reactions, with no evidence of mitochondrial proliferation, but severe deficiencies in SDH activity (figure 2C,G) compared with age-matched control muscle. This was confirmed by the spectrophotometric assay of respiratory chain activities, which demonstrated a severe and isolated deficiency involving complex II in muscle homogenates from both patients (table 1).
Assessment of patient respiratory chain complex activities in skeletal muscle homogenate

(A–C) H&E staining, cytochrome c oxidase (COX) histochemistry and succinate dehydrogenase (SDH) histochemistry respectively, for Patient 1, indicating a marked decrease in the activity of SDH (C) compared with a control SDH reaction (D). (E–H) Images from the muscle biopsy of Patient 2 including H&E staining (E), COX histochemistry reactivity (F), SDH histochemistry reactivity (G) and the Oil Red O stain (H), the last highlighting a very subtle increase in intrafibre lipid.
Molecular genetic analysis
Sequencing of the SDHAF1 gene in both patients revealed no known or potentially pathogenic mutations. Analysis of the coding region of the SDHA gene in Patient 1 revealed two novel heterozygous variants affecting adjacent amino acids within exon 11; a c.1523C>T transition predicting a p.Thr508Ile substitution and a c.1526C>T transition predicting a p.Ser509Leu substitution within the catalytic flavoprotein subunit of complex II (figure 3A). The lack of an SDHA or SDHAF1 defect in Patient 2 prompted analysis of the SDHB gene, identifying a novel homozygous c.143A>T SDHB transversion in exon 2 that predicts a p.Asp48Val substitution within the catalytic iron–sulphur subunit of SDH (figure 3B). Recessive inheritance of all variant alleles was confirmed by screening parental DNA samples (see figure 3A,B). In silico predictions regarding the putative functional effects of the p.Thr508Ile and p.Ser509Leu SDHA and p.Asp48Val SDHB variants supported a deleterious aetiology. Phyre2 and Piccolo modelling revealed no obvious conformational differences between predicted tertiary structures of the wild-type and mutant SDHA and SDHB proteins.

Identification of pathogenic SDHA and SDHB mutations. (A) Compound heterozygous c.1523C>T (p.Thr508Ile) and c.1526C>T (p.Ser509Leu) SDHA mutations were identified in Patient 1, with parental DNA screening supporting recessive inheritance. Both mutations affect highly conserved p.Thr508 and p.Ser509 residues in the SDHA-encoded subunit A of succinate dehydrogenase (SDH). (B) A novel homozygous c.143A>T (p.Asp48Val) SDHB mutation was identified in Patient 2, with recessive inheritance supported by parental DNA screening. Multiple sequence alignment of this region of the SDHB subunit confirms that p.Asp48Val mutation affects an evolutionary conserved residue.
Western blotting
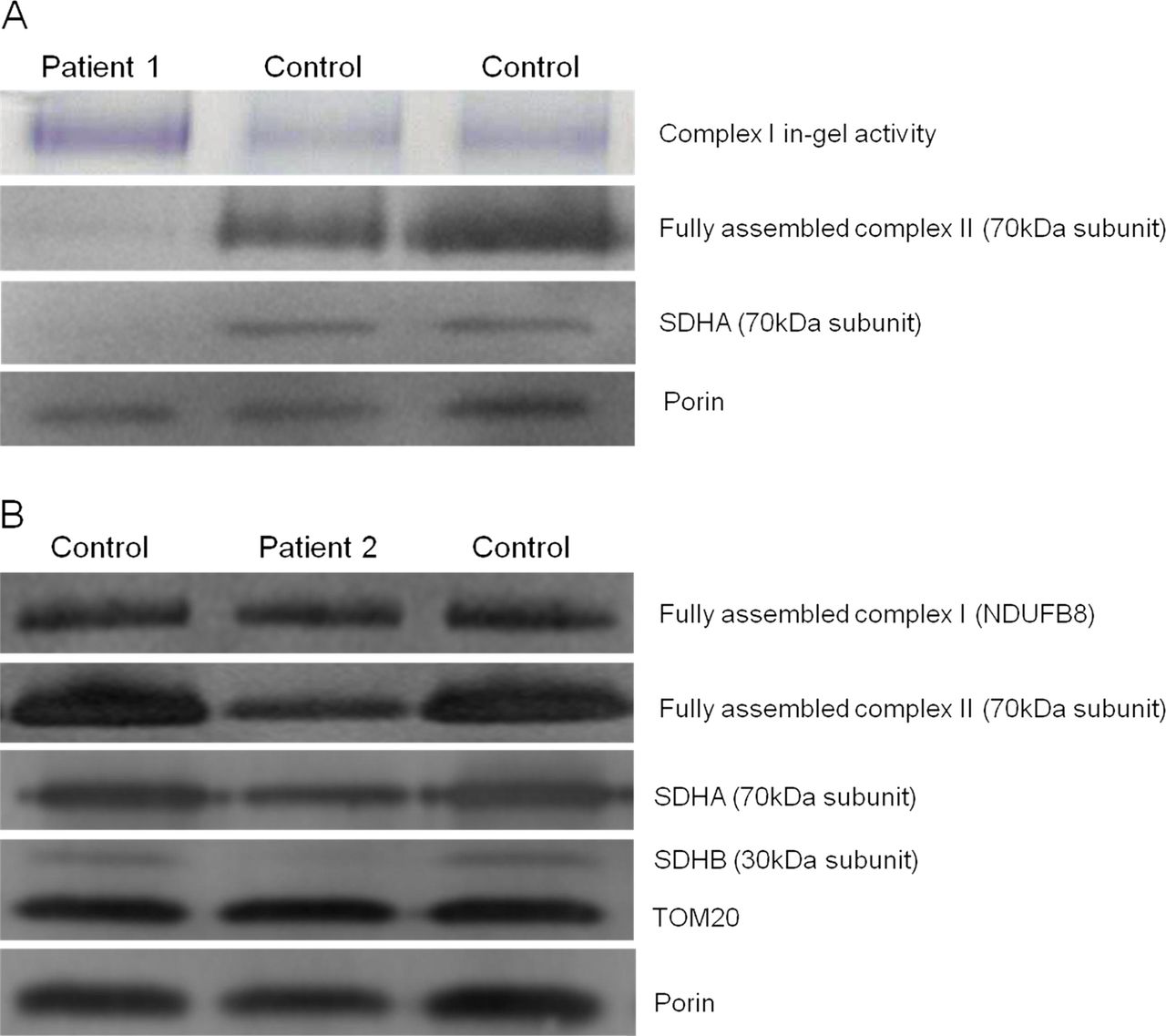
To assess the impact of the novel SDHA and SDHB variants on the steady-state levels of complex II and the respective subunits’ stability, BN-PAGE and SDS-PAGE analyses were performed on available material from the two patients. BN-PAGE analysis using isolated mitochondria from cultured fibroblasts of Patient 1 revealed a significantly decreased amount of fully assembled complex II compared with aged-matched controls (figure 4A). In contrast, comparatively higher complex I in-gel activities were noted in this patient. SDS-PAGE analysis confirmed an almost complete absence of SDHA in the patient, relative to the mitochondrial loading marker, porin. BN-PAGE analysis using enriched skeletal muscle mitochondria from Patient 2 showed a relative lower amount of fully assembled complex II compared with complex I (figure 4B), while SDS-PAGE analysis demonstrated an almost complete absence of the SDHB subunit compared to both control samples and mitochondrial loading controls. In addition, expression of the SDHA subunit of complex II also appeared to be decreased, suggesting instability of the assembled complex.

Investigation of complex activities and protein expression in Patients 1 and 2 and controls. (A) BN-PAGE analysis of mitochondria isolated from cultured patient and control fibroblasts revealed a reduction of assembled complex II. SDS-PAGE analysis, probed with antibodies against porin (loading control) and the flavoprotein subunit of succinate dehydrogenase (SDH), revealed an almost complete obliteration of SDHA expression for Patient 1, corroborating the pathogenicity of the c.1523C>T (p.Thr508Ile) and c.1526C>T (p.Ser509Leu) SDHA variants. (B) BN-PAGE analysis of enriched mitochondria from patient and control muscle revealed a reduction of complex II assembly, with normal complex I assembly. SDS-PAGE analysis shows a gross decrease in iron–sulphur (SDHB) and a slightly reduction in SDHA subunit expression, with normal levels of TOM20 and porin.
Complementation studies in yeast of human SDHB mutant alleles
To validate the pathogenic role of the p.Asp48Val SDHB mutation in Patient 2, we performed complementation studies using the Saccharomyces cerevisae strain BY4741, deleted in the SDH2 gene (the yeast orthologue of mammalian SDHB), hereafter referred to as Δsdh2 strain. The mutated SDHB residue p.Asp48Val is not conserved between human and yeast; Aspartate-48 in human SDHB is conservatively substituted by Asparagine-42 in the yeast Sdh2 protein (figure 5A). We therefore constructed the ‘humanised wild-type’ allele by replacing the codon specific to the amino acid residue of the wild-type SDH2 yeast sequence, with the codon corresponding to the amino acid residue of the wild-type SDHB human sequence. The humanised wild-type SDH2 variant allele (p.Asn42Asp) was then introduced into Δsdh2 yeast mutant and the transformants were tested for oxidative growth on YNB medium supplemented with 2% ethanol or 2% acetate (figure 5B). The ‘humanised allele’ was able to complement the oxidative growth defect of Δsdh2 strain.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Complementation studies in yeast confirming pathogenicity of the p.Asn42Val SDHB mutation. (A) ClustalW alignment of the yeast and human SDHB homologues confirm that although the amino acid sequences are highly conserved, the p.Asp48 residue in human SDHB is not conserved in yeast, with the corresponding SDH2 residue being p.Asn42. (B) Oxidative growth phenotype. The strain BY4741 sdh2Δ was transformed with either pFL38 empty vector, pFL38 carrying the wild-type SDH2, the humanised wild-type (sdh2N42D) allele or the pathological (sdh2N42V) allele. Equal amounts of serial dilutions of cells from exponentially grown cultures (105, 104, 103, 102 and 101 cells) were spotted onto yeast nitrogen base (YNB) plates supplemented with 2% glucose or with 2% ethanol. The growth was scored after 5 days of incubation at 28°C. (C) Respiration was measured in cells grown in YNB supplemented with 0.6% glucose. aAllele carried by the vector introduced into the null Δsdh2 strain; bexpressed as nmol O2/min/mg dry weight. Each value is the mean of three independent experiments. (D) Assessment of succinate dehydrogenase (SDH) activity. Enzyme activity was measured in mitochondria isolated from cells grown to late exponential phase at 28°C in YNB supplemented with 0.6% glucose. The values of the sdh2 mutants are expressed as percentage of the activities obtained in the sdh2Δ/SDH2. Values are means of three independent experiments in duplicate.
We then prepared the second construct by introducing the p.Asn42Val substitution, corresponding to the purported pathogenic mutant residue in the SDHB protein of Patient 2, and measured its effect on oxidative growth, SDH activity and respiration. Growth on 2% ethanol and 2% acetate was barely impaired in transformants carrying the p.Asn42Val substitution (figure 5B). When respiration was measured, both strains sdh2p.Asn42Asp and sdh2p.Asn42Val displayed an oxygen consumption rate equivalent to that of the wild-type strain, suggesting that decreased SDH activity does not impair electron flux through the respiratory chain (figure 5C). We then studied the effect of both the sdh2p.Asn42Val mutation and sdh2p.Asn42Asp humanised wild-type variant on SDH enzyme activity. The results obtained indicated that the SDH activity was reduced by approximately 50% in the strain harbouring the p.Asn42Val mutant allele whereas the SDH activity of the humanised wild-type allele was indistinguishable from that of the parental strain (figure 5D).
Discussion
Mitochondrial complex II is a critical enzyme for cellular respiration and despite its key roles in both the Krebs cycle and the electron transport chain, it represents a rare cause of disease in the general population. Given that so few Krebs cycle enzyme deficiencies are reported, bi-allelic mutations affecting these enzymes appear to be typically incompatible with life; all reported cases are recessive and involve isolated deficiencies of fumarate hydratase, SDH and, most recently, aconitase,39 and are associated with a severe neurological phenotype and very poor prognosis, being almost always fatal in the neonatal period. Conversely, OXPHOS disease presentations are comparatively common, can present at any stage throughout life and are associated with vast clinical and genetic heterogeneity. Pathogenic mutations have been identified in numerous OXPHOS genes, but the correlation with clinical phenotype is rarely pathognomonic. Isolated deficiencies involving SDH are the rarest of all OXPHOS deficiencies, accounting for approximately 2% of all mitochondrial disease cases.
Here, we describe two children who presented during infancy with motor manifestations of leukodystrophy and one who also had significant cardiomyopathy, in whom biochemical and histochemical analyses of respiratory chain activities in skeletal muscle uncovered a severe, isolated deficiency of complex II. Mutations in SDHAF1 and SDHA genes are known to cause mitochondrial complex II deficiency;7 ,10 ,11 thus, sequencing of these genes was prioritised. This analysis revealed novel compound heterozygous variants of unknown pathological significance within the SDHA gene (c.1523C>T; p.Thr508Ile and c.1526C>T; p.Ser509Leu) for Patient 1, while analysis of the SDHAF1 and SDHA genes for Patient 2 revealed only wild-type sequence. A candidate gene sequencing approach led to the discovery of a novel homozygous (c.143A>T; p.Asp48Val) SDHB variant in Patient 2. Recessive inheritance of the SDHA and SDHB variants was confirmed by parental DNA screening. Functional investigations supported the deleterious effect of the putative SDHA and SDHB mutations, with SDS-PAGE, BN-PAGE and western blotting confirming decreased levels of SDHA and SDHB protein expression for Patients 1 and 2, respectively; both patients had a marked reduction in stable, fully-assembled complex II.
While there are numerous reports describing SDHB mutations in the context of hereditary and sporadic cancer pathology, this report represents the first case of SDHB mutation in association with a neurological phenotype. Additional functional evidence supporting the pathogenicity of the novel homozygous SDHB mutation was provided through modelling the p.Asp48Val mutation in Saccharomyces cerevisae Despite a very high degree of conservation across the SDHB/SDH2 genes, a lack of homology at the human p.Asp48 SDHB locus and corresponding Saccharomyces cerevisae p.Asn42 residue was noted and addressed in our experimental design. Site-directed mutagenesis was employed to generate a p.Asn42Val mutant, in which a marked decrease in SDH activity was observed. A humanised wild-type yeast model was subsequently generated to investigate whether the lack of homology at the p.Asn42/p.Asp48 locus impacted upon SDH activity; SDH activity in the humanised p.Asp42 and wild-type Sdh2 model were comparable, thus corroborating the pathogenicity of the human p.Asp48Val mutation.
Based on Piccolo modelling, the wild-type human p.Asp48 residue is not involved in direct binding to other SDH residues but is located within the highly conserved 2Fe-2S binding domain of the SDHB protein. While p.Asp48 has no obvious primary interaction with other residues of the complex, we hypothesise that it is critical for efficient binding and onward procession of electrons within the mitochondrial respiratory chain.
We provide functional evidence linking the primary SDHB defect with specific brain pathology. The abnormal MRI signal intensities in brain are restricted to the white matter and indicative of myelin or glial pathology (ie, leukodystrophy); such anomalies have been described in mitochondrial disease, while the spinal cord changes as seen in Patient 1 have been described in mitochondrial pathologies including leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (LBSL).40 The accumulation of succinate detectable in the occipito-parietal (clinically affected) region by MRS provides further evidence of a metabolic aetiology; elevated succinate on MRS imaging investigations could be a key diagnostic indicator of SDH deficiency with the elevated metabolite levels reflecting ‘enzymatic cascade stalling’ and MRS should be considered in patients with suspected mitochondrial disease. Succinate inhibits prolyl hydroxylase activity, which stabilises hypoxia-inducible factor (HIF1α) under ‘normal’ anaerobic conditions (eg, exercise or high altitude) and triggers hypoxia-inducible pathways. In the case of SDH deficiency, metabolic stalling generates an accumulation of succinate that mimics this process.41 This report provides mechanistic evidence illustrating a link between specific white matter changes and SDH deficiency. The factors determining localisation of these effects to specific brain regions remain unclear.
In addition to their recognised role in cellular respiration, SDHA, SDHB, SDHC, SDHD and SDHAF2 have all been shown to possess tumour suppressor functionality, with loss of heterozygosity of SDH gene germline mutations leading to cellular proliferation in paraganglioma and pheochromocytoma,13–15 ,42 GIST,43 small cell renal carcinoma44 and neuroblastoma.45 The reported penetrance of SDHB mutations is the highest of all SDH genes in familial cases of head and neck paraganglioma and pheochromocytoma, being associated with early-onset tumourigenesis.44 The link between SDH defects and tumourigenesis can be made on more than one level. First, characterised by the Warburg effect, the ability of tumours to switch from aerobic to anaerobic/glycolytic respiration is key for immortalisation.46 Second, succinate-directed activation of the HIF pathway inhibits apoptosis and stimulates angiogenesis.47 ,48 Moreover, there is evidence supporting an increase of reactive oxygen species (ROS) in systems with SDH deficiencies; increased ROS is a recognised cause of DNA damage, further compounding the already mounting oncogenic pressures on the cell, increasing the likelihood of transformation to immortalised cell status.41
Proving the pathogenicity of candidate gene mutations is always important, perhaps more so when the genes involved are also tumour suppressors. Having confirmed the SDH mutation as pathogenic, there are further ethical considerations with regard to disclosure of carrier status and prenatal testing. A child born to carrier parents has a 25% risk of a severe neurological phenotype, a 50% risk of ‘elevated cancer susceptibility’ and in the situation where only one parent harbours a SDHB mutation, a 50% risk of elevated cancer susceptibility. The issue of whether clinicians should be able to reveal carrier status as well as ‘clinically affected’ status during prenatal screening is one that should be considered indepth, particularly if there is evidence of oncogenesis in relation to the SDH gene mutation in question.
While the recessive genetic defects in SDHA and SDHB identified in the two probands described here have caused severe neurological presentations and complex II deficiency, the fact that the parents of Patients 1 and 2 are heterozygous carriers of germline SDH mutations may place them at an elevated risk of tumourigenesis although neither the SDHA (p.Ala508Thr and p.Ser509Thr) nor SDHB (p.Asp48Val) mutations are reported in the Leiden Open Variation Database (http://chromium.liacs.nl/LOVD2/SDH/home.php), suggesting that these particular mutations have not yet been linked to tumourigenesis. Although there is no indication of cancer susceptibility in either family, both families have been referred for surveillance.
As with the majority of mitochondrial disease presentations, there are no effective cures for SDH deficiency although riboflavin has been shown to alleviate some of the symptoms and delay disease progression.49 Following the diagnosis of SDH deficiency, oral coenzyme Q10 treatment was commenced in Patient 2 and a subjective improvement in strength was reported.
We recommend screening of the SDHA, SDHB and SDHAF1 genes for patients with a biochemically and histochemically characterised isolated complex II deficiency. Identifying the underlying genetic basis of isolated complex II deficiency is vital to ensure that appropriate counselling is available for the family. It facilitates access to cascade screening, and given the increased cancer susceptibility, particularly in relation to SDHB defects, routine surveillance would enable early detection of tumours and appropriate intervention.
Acknowledgments
The authors thank Dr Marie-Anne Brundler and Mr Gavin Falkous for their help with the histopathology.
References
Footnotes
-
Contributors CLA, JED, PGo, IF, RM and RWT contributed to the project design, analysis of the data and/or the drafting of the manuscript. JED and EW recruited patients and family members and phenotypically characterised the families. CLA, FHvdW, LH and H-TH-D performed the biochemical, molecular and protein analysis. JED, ACP and PGo performed the MR studies. FM, PGo and IF performed the yeast experiments. RWT supervised the study.
-
Funding This work was supported by grants from the Wellcome Trust (906919), Fondazione Telethon (GGP11011), CARIPLO (2011/0526) and the UK NHS Specialist Commissioners who fund the ‘Rare Mitochondrial Disorders of Adults and Children’ Diagnostic Service in Newcastle upon Tyne (http://www.mitochondrialncg.nhs.uk). JED is supported by a Sparks Clinical Research Training Fellowship. FHvdW was supported by a Newcastle University Research Committee Visiting Professorship bursary and a North-West University (South Africa) research grant. H-TH-D is supported by a Deutsche Forschungsgemeinschaft (DFG) postdoctoral fellowship.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethics approval was provided by the Newcastle and North Tyneside 1 Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.