Article Text

Abstract
Background Osteopetrosis is a life-threatening, rare disorder typically resulting from osteoclast dysfunction and infrequently from failure to commitment to osteoclast lineage. Patients commonly present in infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, and bone marrow failure. In ∼70% of the patients there is a molecularly defined failure to maintain an acid pH at the osteoclast-bone interface (the ruffled border) which is necessary for the bone resorptive activity.
Methods and results In eight patients with infantile osteopetrosis which could be cured by bone marrow transplantation, the study identified by homozygosity mapping in distantly related consanguineous pedigrees a missense mutation in a highly conserved residue in the SNX10 gene. The mutation segregated with the disease in the families and was carried by one of 211 anonymous individuals of the same ethnicity. In the patients' osteoclasts, the mutant SNX10 protein was abnormally abundant and its distribution altered. The patients' osteoclasts were fewer and smaller than control cells, their resorptive capacity was markedly deranged, and the endosomal pathway was perturbed as evidenced by the distribution of internalised dextran.
Conclusions SNX10 was recently shown to interact with vacuolar type H+-ATPase (V-ATPase) which pumps protons at the osteoclast-bone interface. Mutations in TCIRG1, the gene encoding a subunit of the V-ATPase complex, account for the majority of cases of osteopetrosis. It is speculated that SNX10 is responsible for the vesicular sorting of V-ATPase from Golgi or for its targeting to the ruffled border. A mutation in SNX10 may therefore result in ‘secondary V-ATPase deficiency’ with a failure to acidify the resorption lacuna. Determination of the sequence of the SNX10 gene is warranted in molecularly undefined patients with recessive ‘pure’ osteopetrosis of infancy.
- Osteopetrosis
- SNX10
- endosomal/lysosomal pathway
- academic medicine
- haematology (incl blood transfusion)
- immunology (including allergy)
- genetics
- calcium and bone
- cell biology
- genetics
- neuromuscular disease
- molecular genetics
Statistics from Altmetric.com
- Osteopetrosis
- SNX10
- endosomal/lysosomal pathway
- academic medicine
- haematology (incl blood transfusion)
- immunology (including allergy)
- genetics
- calcium and bone
- cell biology
- genetics
- neuromuscular disease
- molecular genetics
Introduction
Osteopetrosis (OP) is a life threatening disease caused by subnormal osteoclast function, with an incidence of 1 in 250 000 births. The disease usually manifests in the first few months of life with macrocephaly and frontal bossing, resulting in a typical facial appearance. Skull changes—a consequence of defective bone remodelling—result in choanal stenosis, concomitant respiratory problems, and feeding difficulties which are the first clinical manifestations of the disease. The expanding bones encroach on nerve foramina leading to blindness, deafness, and facial palsy.1 Complete visual loss occurs invariably in all untreated patients and hearing loss is estimated to affect 78% of individuals with OP.2 Tooth eruption defects and severe dental caries are common.3 Calcium feedback haemostasis is impaired and children with OP are at risk of developing hypocalcaemia, with attendant tetanic seizures and secondary hyperparathyroidism. The most severe complication of OP, limiting survival, is bone marrow insufficiency. The abnormal expansion of cortical and trabecular bone physically limits the availability of medullary space for haematopoietic activity leading to life threatening cytopenia and secondary expansion of extramedullary haematopoiesis at sites such as the liver and spleen.
The molecular basis of OP is known in about 70% of the patients.4 Most of the genes involved in OP in humans are associated with the maintenance of acid pH at the osteoclast-bone interface which is necessary for bone resorptive activity.
We report on the identification of a new OP associated gene in consanguineous families of Palestinian origin.
Patients and methods
Patients
Four patients (4367, 4321, 261002, 5151), three boys and one girl originating from two consanguineous Palestinian families (figure 1), were the subjects of this study. Their clinical and biochemical data are presented in table 1. Briefly, all four had older relatives with OP and all were suspected of having OP at 1–4 months of age because of macrocephaly or failure to thrive due to feeding problems caused by upper airway. Their neurocognitive development was age appropriate. Physical examination revealed increased head circumference, broad open fontanelle, frontal bossing, and hepatosplenomegaly. Hearing was normal. Functional vision was impaired due to the presence of severe optic atrophy, but the retinae were intact (table 1). Laboratory investigation revealed anaemia, thrombocytopenia, and high lactate dehydrogenase (LDH). Bone radiographs showed dense bones with no distinction between outer and inner plates due to extensive encroachment of cortical bone into the medullary space. Brain CT scan disclosed narrowed optic and auditory canals with sclerosis of the semicircular canals. The ethmoidal and mastoid air cells and the sphenoidal sinuses were fully ossified. Two patients underwent bone marrow transplantation at 10 and 19 months and on follow-up had normal growth and development.

Simplified family trees showing the relationship between the patients. Not included are, for example, three healthy and five affected, dead sibs of patient 261002 whose DNA were not available. The patients' symbols are filled. Numbered symbols represent individuals whose DNA samples were available for analysis.
Clinical and haematological findings of the patients
Methods
Linkage analysis of the diseased gene, using DNA of the four patients, was performed with the GeneChip Human Mapping 250K Nsp Array of Affymetrix, as previously described.5 All experiments involving DNA of the patients, their relatives, and healthy controls were approved by the Hadassah Ethical Review Committee.
Osteoclast generation
Human peripheral blood mononuclear leucocytes were separated over Ficoll-Hypaque gradient using standard techniques. The interface layer comprising the peripheral blood mononuclear leucocytes was collected, washed in PBS and resuspended in MEM-α/10% FBS containing 60 ng/ml human M-CSF for 72 h and plated onto 96-well plates (150 000 cells/well) or onto Permanox multi-well slides (used for immunofluorescence and dextran internalisation assay). Monolayers were then washed once and incubated with MEM-α/10% FBS containing 60 ng/ml human M-CSF and 100 ng/ml human RANKL for 14 days. The medium was changed on days 4, 7, and 11. Cells were fixed and stained for TRAP using leucocyte acid phosphatase staining kit (Sigma-Aldrich, St Louis, Missouri, USA).
SNX10 immunostaining
The differentiated osteoclasts were fixed with formaldehyde (4% in PBS, 30 min room temperature), quenched with ammonium chloride (1% w/v in PBS), and permeabilised. They were then blocked with 1% BSA, and stained with an SNX10 antibody (Santa Cruz, California, USA, catalogue no. sc-104657) followed by an RRX coupled secondary antibody. Cells were then mounted in 5% w/v propyl-gallate in 80% glycerol, 100 mM Tris pH 9.0. Images were obtained with Zeiss Axiovert microscope equipped with epifluorescence using the Metamorph software.
Dextran internalisation in osteoclasts
Differentiated osteoclasts were incubated with 200 μg/ml aldehyde fixable Alexa 488-dextran (Life Technologies, Grand Island, NY, USA, catalogue no. D22910) for 18 h and then fixed and observed as described above.
Osteoclastic resorption
Mononuclear fraction of peripheral blood was prepared as for osteoclast generation. Cells were plated onto BD BioCoat Osteologic Bone Cell Culture System chamber slides (150 000 cells/chamber). Monolayers were treated as for osteoclast generation. After 14 days with RANKL the medium was removed, wells were rinsed with water, and a bleach solution (6% NaOCl, 5% NaCl) was added to the wells for 5 min followed by three washes with distilled water. Formation of the resorbed area was examined microscopically; to ensure objectivity the interpretation was performed on unmarked slides.
Results
Because of the similar presentation and the remote consanguinity we searched for shared homozygous regions in the DNA samples of the three pedigree A patients, 5151, 261002, and 4321. The DNA single nucleotide polymorphism (SNP) analysis revealed multiple homozygous regions in each patient, but all three shared a single genomic stretch on chromosome 7:11.46–26.60 Mb (numbering according to Human Genome build 18). As part of a larger project aiming at the delineation of the molecular basis of OP among Palestinian patients from consanguineous families, we noted that patient 4367, who originated from an unrelated family (pedigree B), had a partly overlapping homozygous region on chromosome 7:25.38–34.26 Mb. Within the common 1.22 Mb genomic stretch, all four patients shared an identical haplotype over the encompassed 128 SNP markers (rs2391129-rs1029561). Five protein coding genes were present within this NFE2L3, HNRNPA2B1, CBX3, SNX10 and SKAP2—and although SNX10 emerged as a prime candidate, we determined the sequence of all 38 exons and their flanking intronic sequences. No mutation was detected in NFE2L3, HNRNPA2B1, CBX3, and SKAP2; however, a missense mutation, c.152 G→A, was identified in exon 3 of SNX10. The mutation Arg51Gln (R51Q) was present in a homozygous state in the four patients. We screened both unrelated and distantly related patients for the mutation and identified an additional four patients (4913, 4919, 4437, and 4435) whose clinical data were integrated into table 1. Altogether, eight patients were homozygous for the mutation, five available parents and four sibs were heterozygous for the mutation, and one sib was homozygous for the normal allele (figure 2A–C). We also screened 211 anonymous individuals of the same ethnicity and identified a single carrier for the R51Q mutation.
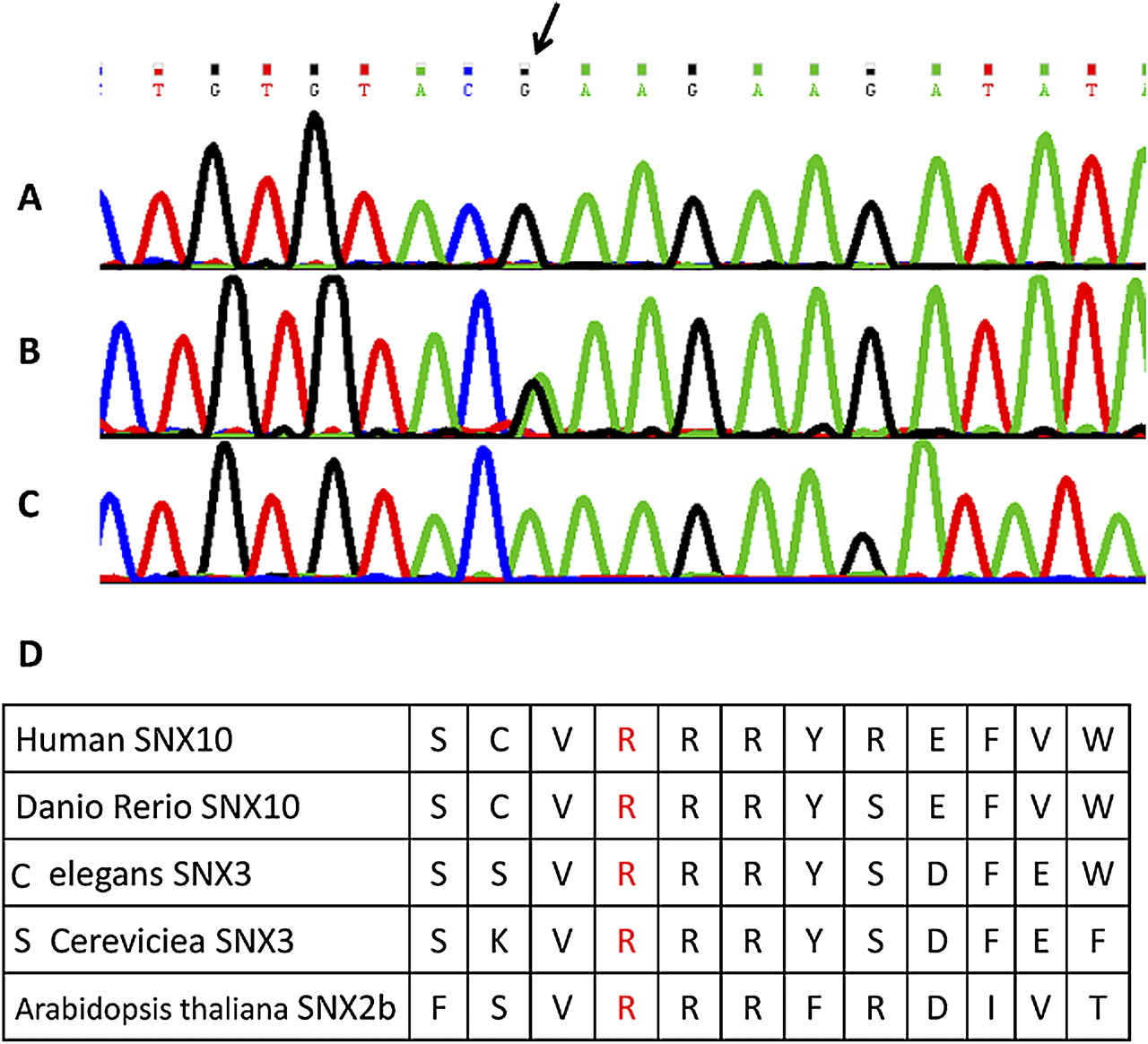
The Arg51Gln mutation in the SNX10 gene. DNA sequence of exon 3 in control (A), obligatory carrier (B), and a patient (C). The mutated nucleotide is indicated (arrow). (D) Conserved amino acid sequence around the altered codon (fourth residue). This figure is produced in colour in the online journal—please visit the website to view the colour figure.
Sorting nexin 10 (SNX10) belongs to a large and diverse group of 33 proteins of the sorting nexin family. Members of this family share a phosphoinositide binding (PX) domain, and are involved in intracellular processes such as endocytosis, protein sorting, and degradation.6 The SNX10 gene consists of six exons encoding 224 amino acids and the PX domain resides in residues 38–135. Arg51 and its surrounding amino acids are conserved throughout evolution (figure 2D) as well as among other simplified members of the sorting nexin family, including SNX3 and SNX11. The substitution of arginine by glutamine at position 51 is predicted disease-causing by the pathogenicity prediction software Mutation Taster, SIFT, and PolyPhen.
In order to study the effect of the R51Q mutation in the SNX10 gene we generated osteoclasts from mononuclear leucocytes of a patient and of an age matched control. Many of the patient cells within the monolayer displayed extensive cytoplasmic vacuolation (figure 3B) which were nearly absent in the control cells (figure 3A). Since SNX10 was proposed to be involved in endosome homeostasis,7 we set out to examine the morphology of the endosomal system in these cells by loading them with fluorescent dextran. This experiment revealed that 25% of the patient cells contained large endosomal vacuoles up to 3–5 μm in diameter (figure 3D). Many of these vacuoles likely corresponded to giant autophagolysosomes since they had a convoluted lumen and accumulated LysoTracker Red (not shown). In control cells (figure 3C) only 2% of the cells displayed these vacuoles, suggesting that mutated SNX10 is associated with abnormal endosomal morphology.

Endosomal and functional abnormalities in osteoclasts derived from mononuclear leukocytes in culture from a patient. Extensive cytoplasmic vacuolation is evident by phase microscopy in patient cells (arrowheads in B) as opposed to control cells (A). These cytoplasmic vacuoles (arrowheads in D) are accessible to internalised dextran Alexa 488 which was used to visualise the endocytic system. Contrasting with these large vacuoles, control cells contain mainly small punctuate staining with dextran (arrows in C). Nuclei are indicated by N in panels A and B. Impaired resorptive function of the patients' cells (E) with fewer and smaller areas cleared from substrate compared to the control (F). Representative fields are shown in the figure.
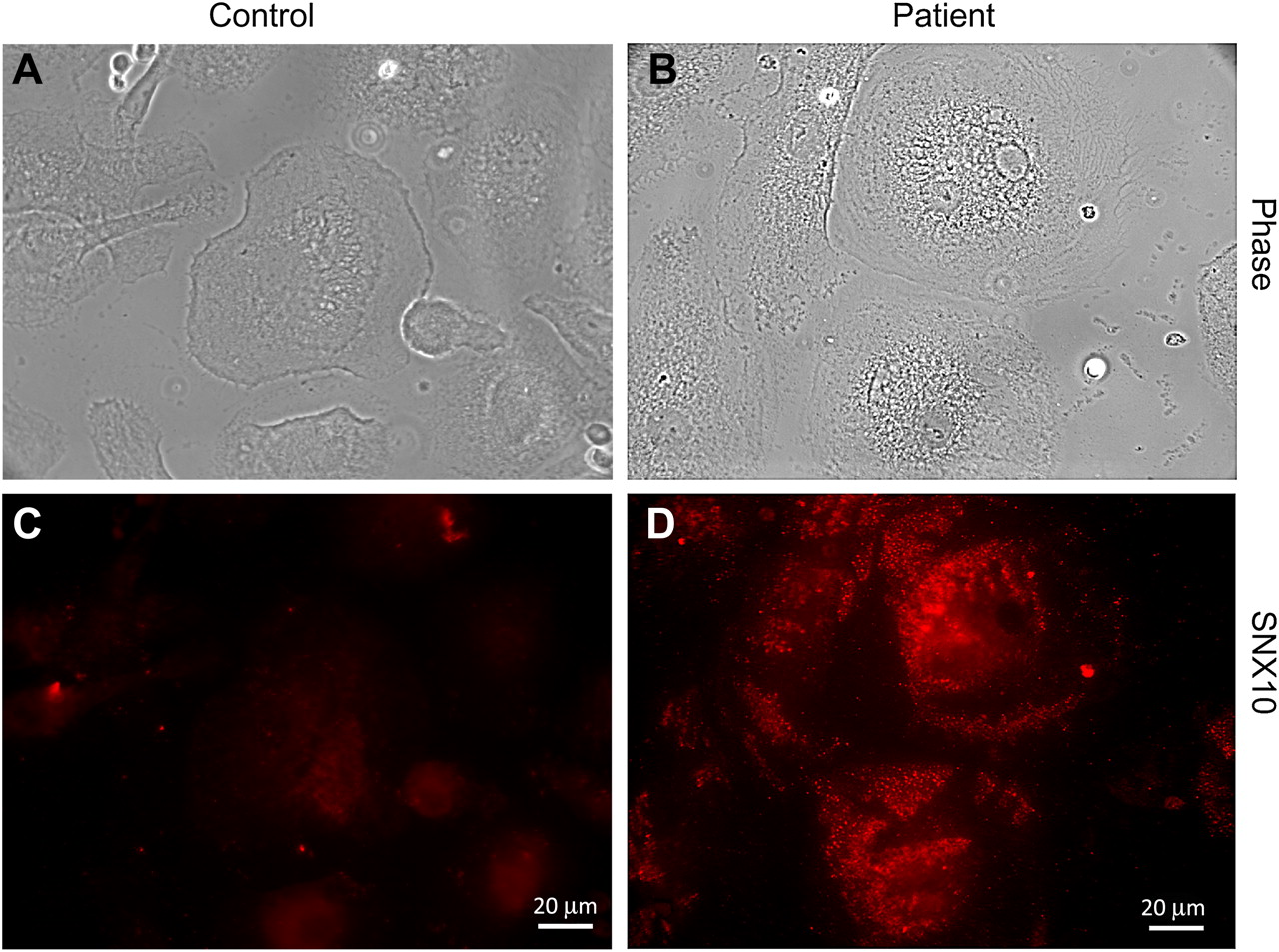
Next we determined the subcellular distribution of the mutant SNX10. Immunostaining of the osteoclasts revealed that the mutant protein in the patient's cells was notably increased. The distribution of the mutant SNX10 was strikingly different from that of the WT protein. Most prominent were SNX10 positive punctae which were clustered throughout the cytoplasm (figure 4C-D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Strikingly different distribution of SNX10 in osteoclast enriched mononuclear leucocytes from a patient. Cells were stained with an SNX10 antibody. In many of the patient's cells (D), SNX10 accumulated in clustered punctae throughout the cytoplasm (compare with control cells in (C)). (A) and (B): phase microscopy of the fields in (C) and (D).
Finally, we studied the resorptive function of the cells using calcium phosphate ceramic thin-film-coated quartz 16-well multitest slides. The patient's cells resorbed far less substrate compared with the control, as revealed by the fewer and smaller areas cleared from substrate (figure 3E-F). It is of note, however, that fewer (∼50–60% of control) and smaller osteoclasts were observed in the patient's derived culture than in the control. It is not clear if this is the result of fewer osteoclast precursors in the patient's peripheral blood or of an impaired differentiation. Taken together, the results suggest that the R51Q mutation in SNX10 is associated with dysfunction and possibly impaired differentiation of osteoclasts, likely by interfering with their endosomal pathway.
Discussion
What might be the role of SNX10 in OP? OP results from failure of osteoclasts to dissolve mineralised bone. Osteoclasts are generated from bone marrow derived monocyte/macrophage precursor cells. An essential signal for initiating osteoclastogenesis is the activation of the receptor activator of nuclear factor-κB (RANK) by its specific ligand, RANKL. RANKL is a haematopoietic factor, carried by osteoblasts and stromal cells, and is involved in the generation of cytosolic Ca2+ oscillations. These are important for the expression of the master regulator of osteoclastogenesis, nuclear factor of activated T cells c1 (NFATc1). Osteoclasts function in bone resorption via their ruffled border, a convoluted membrane within the resorption lacuna. This space is acidified by a vacuolar type H+-ATPase (V-ATPase), a multi-subunit complex which pumps protons generated by carbonic anhydrase II. Electroneutrality is maintained by the chloride channel CLCN7 which transports chloride ions into the lacuna as counter ions. CLCN7 requires transmembrane protein 1 (OSTM1) for its proper localisation. Both V-ATPase and CLCN7 are brought to the ruffled border by late endosome/lysosome, underscoring the importance of vesicular trafficking for osteoclastic resorption. Importantly, SNX10 was recently shown to co-immunoprecipitate with V-ATPase and regulate its intracellular trafficking.8
Mutations in several osteoclast related genes were heretofore associated with OP,4 mostly involving the maintenance of acid pH at the osteoclast-bone interface; about half of the patients carry recessive mutations in TCIRG1 which encode the α-3 subunit of V-ATPase.9 An additional 10–15% of the patients harbour mutations in CLCN7 which encodes the chloride channel. The clinical manifestations of CLCN7 mutations are not only OP but also degeneration of the brain and the retina as a result of abnormal lysosomal storage.10 Rare causes of OP are: mutations in OSTM1 which interacts with CLCN7 and also manifests by neonatal neuro-retinal degeneration and microcephaly, contrasting with the presentation in the more common genetic variants11; mutations in CAII, which encodes carbonic anhydrase type II, and are characterised by the concomitant occurrence of renal tubular acidosis12; and a mutation in Plekstrin homology domain-containing family M member 1 (PLEKHM1), which is operative in vesicular transport in osteoclasts via its interaction with Rab7, a Rab GTPase that localises to the late endosome/lysosome,13 and causes OP because of the osteoclasts' failure to form a ruffled border.14 An additional small group of OP patients suffer from defects in osteoclast differentiation, as a result of mutations in either RANK or its ligand RANKL.15 16
We hereby show that a mutation in a conserved residue in the SNX10 gene is associated with: (1) a strikingly altered cytoplasmic distribution of an excessive amount of the mutant protein; (2) perturbation of the endosomal/lysosomal pathway, demonstrated by the generation of large vacuoles, perhaps autophagolysosomes, which accumulate vast amounts of internalised fluorescent dextran; and (3) abnormally small osteoclasts with reduced resorptive capacity. Since the resorptive capacity of the patients' osteoclasts is decreased and the clinical phenotype is reminiscent of that of patients with TCIRG1 mutations, we speculate that SNX10 is responsible for the vesicular sorting of the V-ATPase complex from the Golgi network or for its targeting to the ruffled border. A mutation in SNX10 would therefore result in a ‘secondary V-ATPase deficiency’ with a failure to acidify the resorption lacuna. In view of the complete cure achieved by allogenic bone marrow transplantation, we suggest that the functional impairment caused by SNX10 mutation is restricted to haematopoietic lineage. Determination of the sequence of the SNX10 gene is warranted in molecularly undefined patients in order to estimate its relative contribution to OP.
Acknowledgments
We thank Judith Melki for linkage analysis and Lital Sheva and Rachel Dahan for excellent technical assistance.
References
Footnotes
Funding This study was supported by the Hadassah-Hebrew University Joint Foundation grant to SH.
Competing interests None.
Ethics approval Ethics approval was provided by Hadassah Helsinki committee.
Provenance and peer review Not commissioned; externally peer reviewed.