Article Text

Abstract
Background The recurrent ∼600 kb 16p11.2 BP4-BP5 deletion is among the most frequent known genetic aetiologies of autism spectrum disorder (ASD) and related neurodevelopmental disorders.
Objective To define the medical, neuropsychological, and behavioural phenotypes in carriers of this deletion.
Methods We collected clinical data on 285 deletion carriers and performed detailed evaluations on 72 carriers and 68 intrafamilial non-carrier controls.
Results When compared to intrafamilial controls, full scale intelligence quotient (FSIQ) is two standard deviations lower in carriers, and there is no difference between carriers referred for neurodevelopmental disorders and carriers identified through cascade family testing. Verbal IQ (mean 74) is lower than non-verbal IQ (mean 83) and a majority of carriers require speech therapy. Over 80% of individuals exhibit psychiatric disorders including ASD, which is present in 15% of the paediatric carriers. Increase in head circumference (HC) during infancy is similar to the HC and brain growth patterns observed in idiopathic ASD. Obesity, a major comorbidity present in 50% of the carriers by the age of 7 years, does not correlate with FSIQ or any behavioural trait. Seizures are present in 24% of carriers and occur independently of other symptoms. Malformations are infrequently found, confirming only a few of the previously reported associations.
Conclusions The 16p11.2 deletion impacts in a quantitative and independent manner FSIQ, behaviour and body mass index, possibly through direct influences on neural circuitry. Although non-specific, these features are clinically significant and reproducible. Lastly, this study demonstrates the necessity of studying large patient cohorts ascertained through multiple methods to characterise the clinical consequences of rare variants involved in common diseases.
- Clinical genetics
- Obesity
- Psychiatry
- Complex traits
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
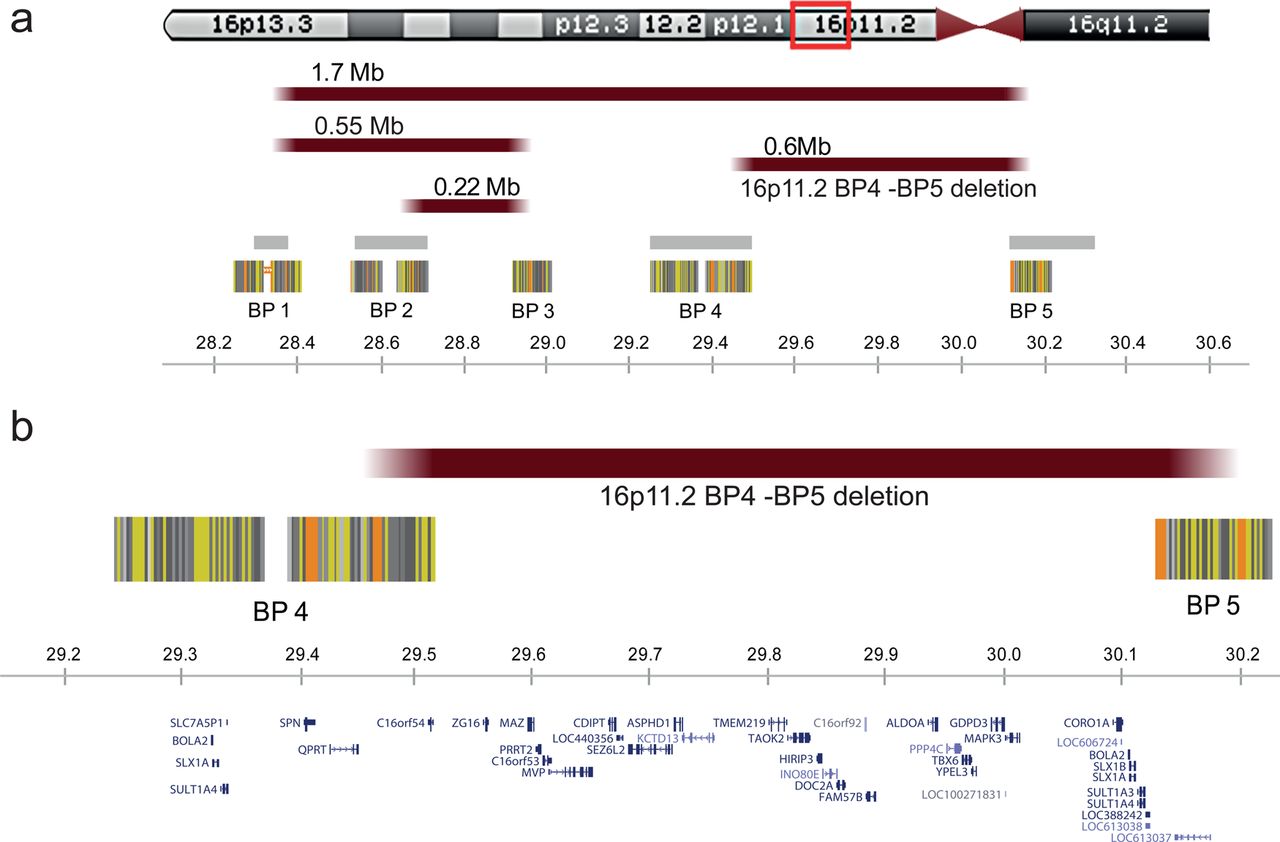
The 16p11.2 locus (figure 1) encompasses several distinct genomic structural variants, including a recurrent interstitial deletion, of a ∼600 kb region containing 29 genes.1 This deletion defined by breakpoints 4 and 5 (BP4-BP5) has a population prevalence of approximately 1/2000,2 and reaches 0.5% in autism spectrum disorders (ASD).3–7 It is one of the most frequent known single locus aetiologies of neurodevelopmental disorders and ASD.8

The 16p11.2 locus. Highly homologous blocks of low copy repeats (LCRs) may act as substrates for non-allelic homologous recombination, predisposing to genomic disorders.48 Five LCRs have been defined as mediators of recurrent and clinically relevant imbalances within the 16p11.2 chromosomal band. To clarify the terminology, we propose to number these ‘recombination hotspots’ from telomere to centromere as breakpoints BP1 to BP5. The current study describes only features associated with the proximal 600 kb recurrent deletion, delineated by BP4 and BP5 at genome sequence coordinates 29.5 and 30.1 Mb, respectively. Distal BP2-BP3 and BP1-BP3 mediated rearrangements, of respectively 220 and 550 kb, containing the SH2B1 gene, have also been reported in individuals with early onset obesity and variable degrees of developmental delay.49 Several recurrent rearrangements overlap the proximal BP4-BP5 region studied here including the 1.7 Mb deletions and duplications from BP1 to BP59 which should be considered as distinct entities. (A) Rearrangements are schematically pinpointed with reddish bars while grey bars and striated blocks indicate intervals of recurrent polymorphisms reported in the Database of Genomic Variants (http://projects.tcag.ca/variation) and common sequence stretches, respectively. (B) Genes encompassed by the genomic region between BP4 and BP5 are shown. All genomic positions are given according to the human genome build hg18/NCBI 36.
We and others have demonstrated that this deletion predisposes to a highly penetrant form of obesity with a 43-fold increased risk of developing morbid obesity.1 ,9 Increased head circumference (HC) has also been associated with the deletion.2 ,10 A mirror phenotype is observed in carriers of the reciprocal duplication who present a high risk of being underweight and microcephalic.2 ,10
The diversity of published clinical features,10–14 together with the report of asymptomatic (but not fully evaluated) transmitting parents,10 ,15 ,16 demonstrated the need to assess systematically the impact of the deletion on neurocognitive development and behaviour.15 To identify the essential clinical traits accurately and assist with genetic counselling, through a collaborative effort we have collected the largest dataset of 16p11.2 BP4-BP5 ∼600 kb deletion carriers. It combines deletion carriers from both the 16p11.2 European and Simons Variation in Individuals Project (Simons VIP) consortia (n=116 and n=52, respectively). We present in this report the natural history, frequency, and range of phenotypes of this rearrangement. We demonstrate that the 16p11.2 deletion consistently impacts cognitive functioning, behaviour, growth, and body mass index (BMI).
Patients and methods
Patients
This study was reviewed and approved by the institutional review board of each site conducting the study. Signed consents were obtained from participants who underwent full assessments. For the data collected through questionnaires, information was gathered retrospectively and anonymously by physicians who had ordered comparative genomic hybridisation (CGH) analyses performed for patient care purposes only.17 Consequently, research based informed consent was not required by the institutional review board of the University of Lausanne which granted an exemption for this part of the data collection.
Carriers were ascertained through several cohorts (table 1). Details on ascertainment and data collection for the participants have been previously published.2 ,18 All participants in the Simons VIP cohort were mapped using whole genome oligonucleotide arrays, and were found to have the common, recurrent 16p11.2 BP4-BP5 deletion. Data for 96 carriers (76 probands and 20 relatives) from the European consortium were obtained by completion of a questionnaire by referring clinicians. Fifty-four probands and 18 relatives (n=72) were extensively evaluated on site by investigators of one of the consortia. Data on 117 deletion carriers ascertained for developmental disorders/intellectual disability (DD/ID) (n=84) , obesity (n=15) and from the general population cohorts (n=18), were collected from our previously published studies, as well as from the literature.1 ,2 ,4 ,5 ,7 ,10 ,11 ,13–16 19–23
Ascertainment of deletion carriers
In the Simons VIP cohort, four patients were excluded based on the presence of additional pathological copy number variants (CNV), other genetic diagnoses, birth asphyxia, fetal alcohol syndrome, and/or prematurity <30 weeks. In the European series, known additional variants (three carriers with a CNV >500 kb and a pair of twins with an FGFR3 mutation) did not represent an exclusion criterion. The inclusion or exclusion of these five carriers did not have an impact on any results (supplementary table S1). They were included in the final analysis because of the arbitrariness of this filtering that only takes into account visible rearrangements and/or known point mutations. We have to assume that many additional mutational events not detectable by CGH array may be present in this dataset.
In the VIP cohort, ethnicity was 75% Caucasian, 5.8% African American, 1.9% Native American, 7.7% other (mixed ancestry), 9.6% unknown (adopted). For the 72 patients evaluated on site in the European cohort, all carriers were of European descent.
All available data on patients ascertained for DD/ID from the 16p11.2 European cohort, Simons VIP and the literature (n=285) were pooled for statistical analysis. Due to missing data for some phenotypes, the denominator changes, which is specified throughout the text. Data collected through questionnaires or by direct assessment are presented separately in supplementary tables S2 and S3. The 33 deletion carriers from the general population and obesity cohorts, for which only anthropometric data were available, are used in the analysis of growth parameters alone. Full scale intelligence quotient (FSIQ) was assessed in 68 intrafamilial controls. The Simons Simplex Collection (SSC)24 was used as an ASD reference population to investigate the effect of obesity on the frequency of macrocephaly in patients with a neurodevelopmental disorder.
Cognitive functioning, psychiatric and behavioural assessment
Overall cognitive functioning was assessed using either the Mullen Scales of Early Learning, the Differential Ability Scales—Second Edition (Early Years and School Age, DAS-II), the Wechsler Intelligence Scales (WPPSI-III; WISC-IV; WAIS-III) or the Wechsler Abbreviated Scales of Intelligence, depending on the age and ability level of the individual.25 Vineland Adaptive Behaviour Scales (VABS)26 were used to measure adaptive skills in daily life. Intellectual disability (ID) was diagnosed as having an FSIQ of 70 or below on a standardised IQ test as well as concurrent deficits in adaptive functioning as defined by the Diagnostic and Statistical Manual of Mental Disorders, 4th edition, text revision (DSM-IV-TR) criteria.27
Different standardised neuropsychological measures were used to assess global cognitive functioning of carriers in the literature.5 ,11 ,15 ,16 ,23 For the purpose of this study, throughout the text we use the term FSIQ to refer to normalised cognitive levels (general population mean=100; standard deviation (SD)=15). ASD was diagnosed by experienced, research-reliable clinicians using the Autism Diagnostic Interview—Revised (ADI-R)28 and the Autism Diagnostic Observation Schedule (ADOS).29 The Diagnostic Interview for Genetic Studies (DIGS)30 was performed for adults, while children's behavioural and emotional problems were assessed with parent report questionnaires.31–33 Additional DSM-IV-TR diagnoses were made by licensed psychologists and psychiatrists using history, parent report and in-person interview. For all aforementioned evaluations, the clinicians were not blinded to the genetic status of the participants.
Statistical analyses
To prevent bias due to intrafamilial correlations, deletion-carrying relatives of probands were excluded as required to avoid including more than one member of the same family in a single analysis. Obesity and morbid obesity in adults are defined throughout the study as BMI ≥30 and 40 kg/m2, respectively. Z scores were computed for all data using gender, age, and geographically matched reference populations as previously described.1 Obesity in children and macrocephaly were defined as BMI and HC Z score ≥2, respectively.
One-tailed Fisher's exact test was used to compare frequencies of the deletion in patients and controls. Two-tailed Student's t test was performed to assess whether BMI, height, weight, and HC Z scores of deletion carriers were different from zero (general population mean). Correlation between these features and FSIQ were examined using Spearman correlation. The binomial test was performed to test for gender bias.
A linear model was applied to correct HC Z scores for BMI, age, and gender effects, using Matlab function regress. A two-tailed Student t test was performed to test the residual effects being different from zero. Linear mixed effect model was used to analyse the longitudinal and cross-sectional data and properly handle autocorrelations. Model fitting was performed to obtain the mean Z score and the corresponding p value for a given time window. The calculations were done using the lme function from the R package nlme.
For each of the W age windows the mean Z scores were computed for each of the P patients resulting in a W×P matrix. Since W tests were carried out, we applied a multiple testing correction that takes into account the correlation structure of the test statistics. As a first step, the effective number of tests (Weff) was derived based on the Pearson's correlation matrix of the W×P dataset.34 We then applied Bonferroni correction of the linear mixed effect model p values, but using Weff instead of W tests to correct for multiple testing.
Breakpoint mapping with short arm of chromosome 16 custom array CGH
To confirm 16p11.2 deletions and ensure that the breakpoints are within the BP4 and BP5 low copy repeats (figure 1), we hybridised Cy3-labelled DNA of the European patients to custom made Nimblegen arrays. These arrays contained 71 000 probes spread across the short arm of chromosome 16 from 22.0 to 32.7 Mb (at a median space of 45 bp between 27.5 and 31.0 Mb) and 1000 control probes situated in invariable region of the X chromosome.2 Cy5-labelled DNA from the GM12042 CEPH (Centre d'Etude du Polymorphisme Humain) cell line was invariably used as reference. DNA labelling, hybridisation and washing were performed according to Nimblegen protocols. Scanning was performed using an Agilent G2565BA Microarray Scanner. Image processing, quality control, and data extraction were performed using the Nimblescan software V.2.5.
Results
The extent of the 16p11.2 rearrangements was assessed by aCGH through standard medical diagnostic procedures and confirmed by a high density custom made array as published (see Methods).2 ,35 Only carriers of the ∼600 kb 16p11.2 BP4-BP5 deletion were considered for further phenotyping; we gathered clinical information on 285 such carriers (figure 1). Cognitive functioning was evaluated in 71 carriers (mean FSIQ=76.1; SD=16.4; figure 2A) and was on average 32 points lower in de novo carriers when compared to their non-carrier family members (∼−2 SD, p=3.96×10−27; table 2A). There is a trend towards higher FSIQ in de novo (n=32) versus inherited (n=14) carriers (FSIQ 83 vs 74; p=0.13; table 2A and supplementary table S1). Of note, parents of de novo deletion carriers who directly enrol their child through a web based questionnaire (VIP cohort) have a higher IQ than the mean of the general population. Among carriers, 20% met DSM-IV-TR criteria for ID (65% mild FSIQ 55–70 and 35% moderate FSIQ 40–55). There are no differences in FSIQ between probands referred for neurodevelopmental disorders and carriers who were not medically ascertained (relatives who carry the deletion) (table 2, supplementary table S1). Carriers’ verbal IQ (mean=74; SD=17.5; range 23–107; n=42) is significantly lower than non-verbal IQ (mean=83.3; SD=18.0; range 47–160; n=43) (p=0.02). Of the 24 carriers evaluated in Europe, 20 (83%) had speech and language therapy during childhood.
FSIQ (A), behavioural and psychiatric features (B) in deletion carriers and intrafamilial controls

Distribution of full scale intelligence quotient (FSIQ) and body mass index (BMI) in deletion carriers.(A) Distribution of FSIQ of 16p11.2 BP4-BP5 deletion carriers (grey bars), intrafamilial non-carrier relatives (control, blue bars) and general population (blue bell curve). The red dashed vertical line represents the FSIQ threshold (70) for intellectual disability (ID). FSIQ is on average 32 points lower in carriers (n=71; mean=76.1; SD=16.4) when compared to their relatives who did not carry the deletion (n=68; mean=108.3; SD=10.9). SD in carriers is similar to that of the reference population (mean=100; SD=15). Bin size was calculated to obtain 10 equal sized bins. (B) Cross-sectional distribution of BMI in carriers (circles: female; open squares: male). BMI progressively increases throughout childhood and adulthood. 70% of the adult carriers are obese (BMI ≥30). The dashed lines represent the 3rd and 97th Center for Disease Control and Prevention (CDC) centile, while the dotted lines pinpoint the thresholds for underweight (BMI=18.5), obesity (30), and morbid obesity (40).
The 16p11.2 BP4-BP5 deletion has been repeatedly associated with ASD, and is one of its most frequent known aetiologies.4–8 Of the fully assessed carriers, ∼15% (8/55) of the children and no adults met criteria for ASD by ADOS and ADI-R. More than 70% (51/70) of non-ASD carriers were found to have other DSM-IV-TR diagnoses27 including attention deficit and disruptive behaviour disorders, anxiety disorders, mood disorders, and substance related disorders (table 2B). There is a significant excess of males among carriers ascertained for neurodevelopmental disorders (138 M/75 F; p=2.4×10−4) in contrast to other criteria (table 1). Gender, however, is not a significant covariate of FSIQ, adaptive level (Vineland), behavioural scores (Social Responsiveness Scale, SRS), neurological symptoms or any other trait (supplementary table S1).
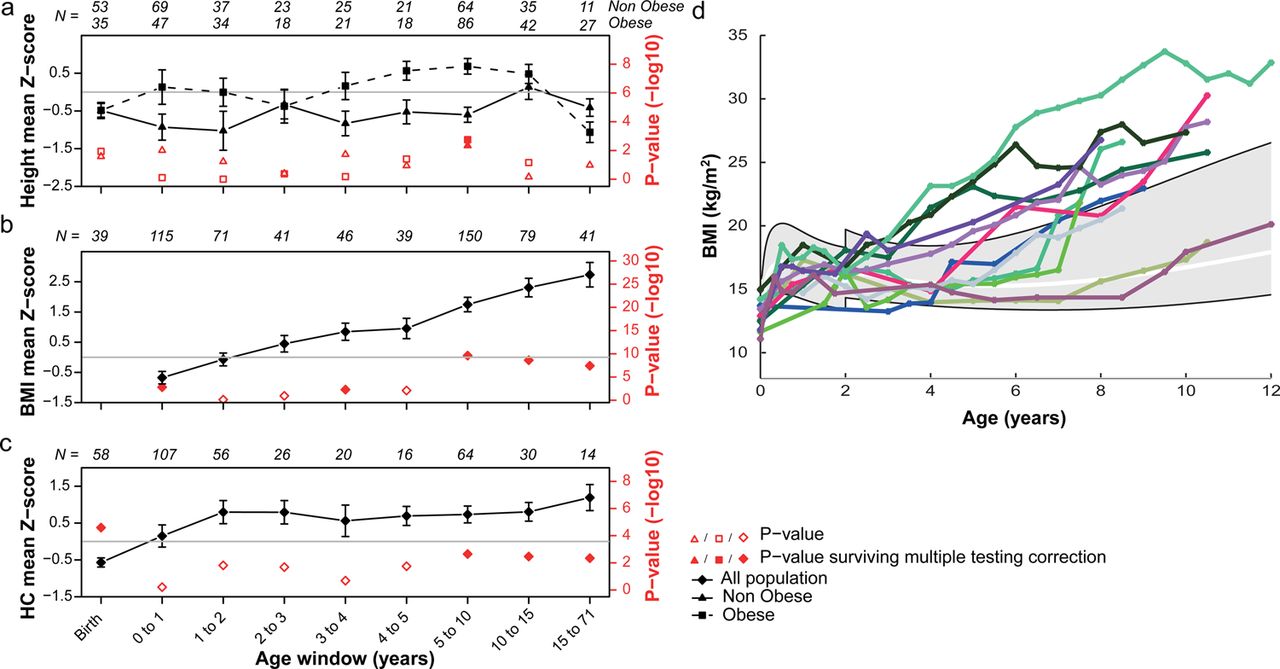
We observed characteristic anthropometric patterns in deletion carriers. Birth weight is below average (n=124, Z score=−0.61; supplementary table S4), whereas Z scores for BMI are significantly higher by the age of 3.5 (Z score=1.01, p=0.03) (figure 3B). Longitudinal data show that this increase in BMI can be sudden and dramatic (figure 3D, supplementary figure S1). By the age of 7 years, obesity is a major comorbidity present in more than 50% of the carriers (supplementary figure S2). Excluding carriers ascertained for obesity, the frequency reaches 75% in adults (36/48), and among all adult obese patients 45% are morbidly obese (figure 2B). The variance of BMI in deletion carriers is higher than in the general population (p=2.06×10−8). Hyperphagia was recorded (through parental questionnaires or reports) in all carriers (n=14) with obesity examined at the European site. The most severe cases require strict control of food access through locked cupboards and refrigerator.

{kind=link}
{kind=link}
{kind=link}
Height, body mass index (BMI), and head circumference (HC) in 16p11.2 BP4-BP5 deletion carriers through development. Height (panel A), BMI (panel B) and HC (panel C) mean Z scores (and corresponding p values in red) for each age window were computed using a mixed effect model to analyse longitudinal and cross-sectional data together. p Values are derived from a two-sided t test of the fixed effects estimates probing whether they are significantly different from 0. Full red dots are p values surviving multiple testing correction (significance's threshold at 6.3×10−3 for height in both obese and non-obese, at 5.6×10−3 for BMI, and at 7.1×10−3 for HC) as opposed to empty red dots. Number of cases N is indicated for each age category. Panel A: Deletion carriers were classified in two groups; either the ‘obese group’ (squares) if they presented obesity at least once during their development, or the non-obese group (triangles). Height is significantly increased in prepubertal obese carriers while non-obese children remain slightly shorter than the general population. Panel B: BMI is significantly elevated by 3.5 years of age. Panel C: HC follows a rapid increase (+1.74 Z score, p=4.8×10−4) during infancy, and remains high throughout life. Panel D: Longitudinal measures of BMI in a subset of 12 carriers illustrating different age onsets of BMI acceleration. The grey area specifies the interval between the 3rd and 97th centile as defined by the WHO data (http://www.who.int/childgrowth/en) between 0–2 years and the Centre for Disease Control and Prevention data above 2 years of age. The white line marks the 50th centile. All available longitudinal data are included in supplementary figure S2.
BMI correlates neither with FSIQ (supplementary table S5) nor behavioural or adaptive scores (SRS, VABS). Furthermore, it is independent of ascertainment methods, inheritance, gender, or the presence of neurological features (supplementary table S1). Childhood obesity is, however, often associated with accelerated prepubertal linear growth, although the mechanism underlying this phenomenon remains unclear.36 In obese deletion carriers, a similar association is observed (figure 3A) in children whose height is increased (n=57, Z score=0.54, p=6.8×10−4) and peaks at 9 years of age (Z score=1.08, p=1.5×10−3). This is to be contrasted with short stature, which is apparent in probands (n=182, Z score=-0.33, p=3.2×10−3), newborns (n=88, Z score=−0.49, p=1.3×10−3), non-obese children (n=91, Z score=−0.59, p=4.4×10−4), and adults (n=32, Z score=−1.1, p=8.0×10−5) (figure 3A, supplementary tables S1, S4, supplementary figure S3).
An overall increase in HC is observed in probands (n=146, Z score=0.56, p=4.0×10−4) with macrocephaly (HC Z score ≥2) present in 29/170 (17%) of the carriers. HC correlates positively with BMI (r=0.45, p=2.8×10−8) (supplementary table S5) and remains elevated after correcting for BMI, age, and gender (linear model; mean corrected Z score=0.60, p=1.1×10−5). Obese deletion carriers show an increased frequency of macrocephaly when compared to non-obese carriers (32% and 10%, respectively; p=1.8×10−3). Longitudinal data (figure 3B,C, supplementary figure S4) show that HC, which is lower at birth by 0.57 Z scores (n=58), increases during infancy (+1.74 Z scores, p=4.8×10−4), several years before the elevation of BMI Z scores. In the Simon Simplex Collection of ASD children, 41.3% of patients with obesity have macrocephaly when compared to 12.5% in non-obese patients (p=5.4×10−26) (supplementary figure S5).
Neurological features observed in patients referred for DD/ID include a wide range of findings (supplementary table S2). Gross motor delay was reported in 37.6% of the patients (32/85 ascertained for DD/ID in the European cohorts), which is consistent with previous series,10 ,23 ,37 and mean age of walking is significantly delayed (mean=20.5 months, SD=8.6, n=30, p=8.63×10−6). Epilepsy is a frequent feature reported in 47/195 (24%) of the probands. While this frequency is not significantly different in non-medically ascertained carriers (5/38, 13%, p=0.2), it is higher in the fully assessed probands (22/54, 41%, p=1.6×10−2). A small fraction of carriers (12/233) received a diagnosis of paroxysmal dyskinesia syndrome (OMIM 128200). MRI performed in a subset of carriers (n=65) showed mostly mild and non-recurrent features. Posterior fossa and/or craniocervical junction related abnormalities (eg, Chiari I malformation, cerebellar tonsillar ectopia, platybasia) were present in 19 of 41 fully reviewed MRIs.
Malformations or major medical problems are present in 76/130 (58%) of the probands from both cohorts, of which 53 have only a single pathological feature (supplementary tables S3 and S6). In non-medically ascertained carriers identified in the families through cascade testing, this frequency is much lower (10/38, 26.3%, p=7.6×10−4). No specific recurrent malformation sequence or multisystemic involvement is observed (supplementary table S6). There are, however, features likely to be associated with the deletion which, in most cases, do not require treatment such as vertebral abnormalities (hemivertebrae or kyphoscoliosis affect ∼20% of carriers). Facial dysmorphia was visible in 69/150 (46%) carriers, yet no recurrent facial gestalt was observed in either cohort.
The frequency of this BP4-BP5 deletion (table 1) among patients referred for clinical chromosome hybridisation microarray is similar for both consortia (0.28%). Though the two cohorts were assembled in different continents and healthcare systems, as well as using different ascertainment criteria, they share striking similarities with the exception of the rate of inherited cases. Among Simons VIP patients, the deletion arose almost exclusively de novo (table 1). Globally, for participants with available parental data (51%), the deletion occurred de novo in 92/145 cases (including two mosaic cases) (64%) and was inherited in the remaining cases (36%). Assuming a stable prevalence of the deletion, this high rate of de novo events implies decreased fitness of this deletion (0.52 and 0.20 in the European and Simons VIP cohorts, respectively).
Discussion
Our study demonstrates that the recurrent 600 kb BP4-BP5 deletion, which has a prevalence of ∼0.05% in the general population,2 has a consistent impact on traits affecting adaptive skills. FSIQ is decreased by ∼2 SD in carriers of both genders, about 20% of them meeting criteria for ID. Since the FSIQ variance remains identical to that of the general population (p=0.25), approximately 30% of deletion carriers fall within the normal range (FSIQ ≥85). Bias in these estimates is unlikely since there were no differences in FSIQ scores of probands and non-medically ascertained carriers.
FSIQ does not capture the full range of disabilities experienced by deletion carriers. A history of speech therapy is frequent (83%) and psychiatric comorbidities affect >80% of carriers. The penetrance of ASD is 15% in our cohorts (table 2B), supporting the association of this deletion with ASD.38 Increased growth velocity of HC during infancy recapitulates the well documented pattern in idiopathic ASD. This shared head growth pattern, which has attracted considerable attention as a marker of abnormal brain development, has been linked to an increase in white matter volume, brain weight and numbers of neurones in the prefrontal cortex of ASD patients.39
Obesity is a major comorbidity of 16p11.2 deletion carriers, with a penetrance among adults of >70%. Durable weight loss in adolescents or adults has not yet been documented: (1) two adults treated by bariatric surgery relapsed several years later; and (2) an adolescent dropped from 39.5 to 27.7 kg/m2 by dieting and engaging in intense physical activity, but returned to his initial weight 2 years after discontinuing this regimen. Weight control is therefore recommended although no data are available on the efficacy of early intervention in deletion carriers. The reported association of ID with obesity40 has led to the proposal that impaired cognition may result in abnormal eating behaviour and obesity.41 In 16p11.2 deletion carriers, however, obesity occurs independently of cognitive function, adaptive or social behaviour scores.
We hypothesise that the deletion directly affects the neural circuitry involved in all these phenotypes, including energy balance. The early increase in HC precedes the onset of obesity (supplementary figure S4). This increase in HC is also observed later in childhood and adolescence (figure 3C) when correlation with brain volume is lower and contribution of skull thickness higher.42 ,43 Furthermore we demonstrate that childhood obesity in 16p11.2 deletion carriers, as well as in patients with autism (SSC cohort), is a confounding factor contributing to increased HC (supplementary figure S5). This deletion lowers final adult height by 1 SD (supplementary figure S3), though the well documented association between idiopathic childhood obesity and prepubertal height acceleration36 is maintained in obese children carrying the deletion (figure 3A,B, supplementary figure S3).
Epilepsy is the most frequent neurological disorder observed in deletion carriers, and electroencephalogram (EEG) evaluation should be prescribed if abnormal movements or behaviours suspicious for seizures are observed. A smaller and possibly underestimated fraction of carriers have paroxysmal dyskinesia syndrome. We failed to observe a difference in the presence or absence of epilepsy when stratifying for either cognitive functioning, BMI or HC (supplementary table S1). It is possible that epilepsy and the latter phenotypes are related to haploinsufficiency of distinct genes (figure 1B). Mutations in PRRT2, a gene mapping to the deleted interval, were recently identified in patients diagnosed with epilepsy and paroxysmal dyskinesia,44 whereas a 118kb deletion that encompasses MVP, CDIPT, SEZ6L2, ASPHD1, and KCTD13 segregated in a threegeneration pedigree with ASD and other neurodevelopmental abnormalities but not epilepsy.45 Of note, morpholino-driven reduction of the expression level of the KCTD13 ortholog resulted in macrocephaly in zebrafish, while its depletion in the brain of mouse embryos resulted in an increase of proliferating cells.35
Although we report a large spectrum of malformations (supplementary table S3), the 16p11.2 deletion should not be regarded primarily as a malformation syndrome. Indeed, the majority of these abnormalities are infrequent, suggesting either fortuitous associations or low penetrance (eg, coloboma/microphthalmia) possibly through unmasking of recessive mutations. Vertebral and spinal related anomalies (∼20%, supplementary table S3) seem, however, to be strongly associated with the deletion, suggesting that spine x-ray and orthopaedic evaluations should be routinely performed in deletion carriers. TBX6, which maps within the interval, is a candidate gene for vertebral malformations since mice homozygous for a Tbx6 mutation showed rib and vertebral body anomalies.46 Additionally, TBX6 polymorphisms were associated with congenital scoliosis in the Han population.47
In conclusion, our study demonstrates in two independent datasets that the 16p11.2 BP4-BP5 600 kb deletion consistently and quantitatively impacts cognitive functioning, HC, BMI, and growth. A range of behavioural disorders affects the vast majority of carriers. Probands referred for neurodevelopmental disorders or morbid obesity and carriers who were not medically ascertained (relatives of a proband) show similar values for FSIQ and BMI, suggesting that ‘asymptomatic’ carriers are uncommon. Five carriers within our cohorts presented an FSIQ >100 and may therefore represent the higher end tail of the FSIQ distribution of deletion carriers. All five presented language disorders, autism, disruptive behaviour or obesity. The deleterious impact of the deletion is further highlighted by its low fitness reflected by the rarity of multigenerational carrier families. In contrast to BMI, the variance of global cognitive functioning (FSIQ) is the same among carriers and control population, suggesting that the factors determining its variability are identical to those at play in the general population and unrelated to the 16p11.2 locus.
This comprehensive study of the 16p11.2 BP4-BP5 phenotype helps to guide clinical monitoring and counselling of patients and families and to potentially overcome the genetic counselling challenge posed by its variability. It illustrates that the study of rare variants causing common diseases lacking pathognomonic features requires the assembly and detailed clinical characterisations of large cohorts, recruited using multiple ascertainment criteria.
Acknowledgments
We thank the participants, families, and referring providers for their contribution. SJ is recipient of a ‘bourse de relève académique de la Faculté de Biologie et Médecine de l'Université de Lausanne’ and KM is a grantee of a scholarship from the Swiss Scientific Exchange NMS Programme. This work is supported by the Leenaards Foundation Prize (SJ and AR), the Swiss National Science Foundation (AR and JSB) and a specific SNSF Sinergia grant (AR). Phenotyping of EGC UT individuals was supported by Targeted Financing from Estonian Government grant SF0180142Cs08, Centre of Translational Genomics grant SP1GVARENG, and by the European Union through the European Regional Development Fund, in the frame of Centre of Excellence in Genomics. The Simons VIP work is supported by the Simons Foundation Autism Research Initiative (SFARI). We thank the coordinators and staff at the Simons Simplex Collection (SSC) sites. We are grateful to all of the families at the participating Simons Simplex Collection (SSC) sites, as well as the principal investigators (A Beaudet, R Bernier, J Constantino, E Cook, E Fombonne, D Geschwind, R Goin-Kochel, E Hanson, D Grice, A Klin, D Ledbetter, C Lord, C Martin, D Martin, R Maxim, J Miles, O Ousley, K Pelphrey, B Peterson, J Piggot, C Saulnier, M State, W Stone, J Sutcliffe, C Walsh, Z Warren, E Wijsman). We appreciate obtaining access to phenotypic data on SFARI Base. Approved researchers can obtain the SSC population dataset described in this study by applying at https://base.sfari.org
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
FZ, EHS, NDB, EH: equally contributing first authors.
AR, JSB, WKC, SJ: equally contributing senior authors.
-
Collaborators List of contributors to the 16p11.2 European Consortium: Marie Claude Addor; Benoit Arveiler; Marco Belfiore; Frédérique Bena; Laura Bernardini; Patricia Blanchet; Dominique Bonneau; Odile Boute; Patrick Callier; Dominique Campion; Jean Chiesa; Marie Pierre Cordier; Jean Marie Cuisset; Albert David; Nicole de Leeuw; Bert de Vries; Gérard Didelot; Martine Doco-Fenzy; Bénédicte Duban Bedu; Christèle Dubourg; Sophie Dupuis-Girod; Christina R. Fagerberg; Laurence Faivre; Florence Fellmann; Bridget A. Fernandez; Richard Fisher; Elisabeth Flori; Alice Goldenberg; Delphine Heron; Muriel Holder; Juliane Hoyer; Bertrand Isidor; Sylvie Jaillard; Philippe Jonveaux; Sylvie Joriot; Hubert Journel; Frank Kooy; Cédric le Caignec; Bruno Leheup; Marie-Pierre Lemaitre; Suzanne Lewis; Valérie Malan; Michèle Mathieu-Dramard; Andres Metspalu; Fanny Morice-Picard; Mafalda Mucciolo; Eve Oiglane-Shlik; Katrin Ounap; Laurent Pasquier; Florence Petit; Anne Philippe; Ghislaine Plessis; Fabienne Prieur; Jacques Puechberty; Evica Rajcan-Separovic; Anita Rauch; Alessandra Renieri; Claudine Rieubland; Caroline Rooryck; Katharina Magdalena Rötzer; Mariken Ruiter; Damien Sanlaville; Stéphanie Selmoni; Yiping Shen; Vanessa Siffredi; Jacques Thonney; Louis Vallée; Ellen van Binsbergen; Nathalie Van der Aa; Mieke M. van Haelst; Jacqueline Vigneron; Catherine Vincent-Delorme; Disciglio Vittoria; Anneke T Vulto-van Silfhout; Robert M Witwicki; Simon A. Zwolinski. List of contributors of the Simons VIP Consortium: Alexandra Bowe; Arthur L Beaudet; Christie M Brewton; Zili Chu; Allison G Dempsey; Yolanda L Evans; Silvia Garza; Stephen M Kanne; Anna L Laakman; Morgan W Lasala; Ashlie V Llorens; Gabriela Marzano; Timothy J Moss; Kerri P Nowell; Monica B Proud; Qixuan Chen; Roger Vaughan; Jeffrey Berman; Lisa Blaskey; Katherine Hines; Sudha Kessler; Sarah Y Khan; Saba Qasmieh; Audrey Lynn Bibb; Andrea M Paal; Patricia Z Page; Bethanny Smith-Packard; Randy Buckner; Jordan Burko; Alyss Lian Cavanagh; Bettina Cerban; Anne V Snow; LeeAnne Green Snyder; Rebecca McNally Keehn; David T Miller; Fiona K Miller; Jennifer Endre Olson; Christina Triantafallou; Nicole Visyak; Constance Atwell; Marta Benedetti; Gerald D Fischbach; Marion Greenup; Alan Packer; Polina Bukshpun; Maxwell Cheong; Corby Dale; Sarah E Gobuty; Leighton Hinkley; Rita J Jeremy; Hana Lee; Tracy L Luks; Elysa J Marco; Alastair J Martin; Kathleen E McGovern; Srikantan S Nagarajan; Julia Owen; Brianna M Paul; Nicholas J Pojman; Tuhin Sinha; Vivek Swarnakar; Mari Wakahiro; Hanalore Alupay; Benjamin Aaronson; Sean Ackerman; Katy Ankenman; Jenna Elgin; Jennifer Gerdts; Kelly Johnson; Beau Reilly; Dennis Shaw; Arianne Stevens; Tracey Ward; Julia Wenegrat; Timothy PL Roberts
-
Contributors All authors contributed to the concept, design, acquisition of data or analysis and interpretation of data. All authors contributed to drafting or revising the content and approved the final version.
-
Competing interests Competing interests are disclosed in the ICMJE conflit of interest forms.
-
Ethics approval This study was approved by the IRB of the University of Lausanne, the IRB of the Simons Foundation as well as the IRB of each site conducting the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Data on the Simons VIP subjects are available through SFARI Base at https://sfari.org/resources/sfari-base.
Linked Articles
- Corrections