Article Text

Abstract
Background Joubert syndrome (JBTS) is a predominantly autosomal recessive disorder characterised by a distinctive midhindbrain malformation, oculomotor apraxia, breathing abnormalities and developmental delay. JBTS is genetically heterogeneous, involving genes required for formation and function of non-motile cilia. Here we investigate the genetic basis of JBTS in 12 French–Canadian (FC) individuals.
Methods and results Exome sequencing in all subjects showed that six of them carried rare compound heterozygous mutations in CC2D2A or C5ORF42, known JBTS genes. In addition, three individuals (two families) were compound heterozygous for the same rare mutations in TMEM231(c.12T>A[p.Tyr4*]; c.625G>A[p.Asp209Asn]). All three subjects showed a severe neurological phenotype and variable presence of polydactyly, retinopathy and renal cysts. These mutations were not detected among 385 FC controls. TMEM231 has been previously shown to localise to the ciliary transition zone, and to interact with several JBTS gene products in a complex involved in the formation of the diffusion barrier between the cilia and plasma membrane. siRNA knockdown of TMEM231 was also shown to affect barrier integrity, resulting in a reduction of cilia formation and ciliary localisation of signalling receptors.
Conclusions Our data suggest that mutations in TMEM231 cause JBTS, reinforcing the relationship between this condition and the disruption of the barrier at the ciliary transition zone.
- Developmental
- Genetics
- Neurology
- Clinical genetics
Statistics from Altmetric.com
Joubert syndrome (JBTS [MIM213300]) is a predominantly autosomal recessive disorder characterised by ocumolotor apraxia, abnormal breathing, ataxia and variable developmental delay or intellectual impairment (reviewed in Sattar et al).1 A cardinal sign of JBTS is the presence of a complex midhindbrain malformation consisting of hypoplasia of the cerebellar vermis, abnormally deepened interpeduncular fossa at the level of the upper pons, and elongated and thickened superior cerebellar peduncles. This malformation takes the appearance of a molar tooth on MRI. Extraneurological manifestations, including retinopathy, renal cysts and polydactyly, are present in a subset of affected individuals. JBTS is genetically heterogeneous, with 17 genes described to date,1–13 all of which appear to play a role in the development and/or function of non-motile cilia.
There is a high prevalence of JBTS in the French–Canadian (FC) population of the Lower Saint-Lawrence region of Quebec. We recently performed exome sequencing in 15 individuals (11 families) with JBTS from that region and found that mutations in C5ORF42 explain JBTS in nine individuals (seven families).12 In addition, we identified pathogenic compound heterozygous mutations in CC2D2A, a previously known JBTS gene, in two affected individuals from two different families. The genetic basis of JBTS remained unexplained in four individuals (two families) from this initial study. Here, we follow-up on our previous investigation by performing exome sequencing in eight additional individuals with JBTS (six unrelated families) originating from other regions of Quebec.
The six probands had a molar tooth sign on imaging, and variable expression of the classical JBTS features. The two additional individuals are the uncle (II-4) and aunt (II-6) of subject IV-1 in family 385/447. These individuals were considered to have a variable expression of JBTS as they both had oculomotor apraxia and, additionally, II-4 had gait ataxia and a history of developmental delay. Brain MRI was normal in II-6 (see online supplementary figure S1, A-B) but was not done in II-4. Informed consent was obtained from each individual or legal guardian. This study was approved by our institutional ethics committee. Genomic DNA from each sample was captured with the Agilent SureSelect 50 Mb exome capture oligonucleotide library, and the captured DNA was sequenced with paired-end 100 bp reads on Illumina HiSeq 2000 resulting in an average of 12.7 gigabases (Gb) of raw sequence for each sample. Data were analysed as previously described.14 After removing putative PCR-generated duplicate reads using Picard (V.1.48), we aligned reads to human genome assembly hg19 using a Burroughs–Wheeler algorithm (BWA V.0.5.9). Median read depth of bases in consensus coding sequence (CCDS) exons was 99 (determined with Broad Institute Genome Analysis Toolkit V.1.0.4418).15 On average, 87% (±2.0%) of bases in CCDS exons were covered by at least 20 reads. We called sequence variants using Samtools (V.0.1.17) mpileup and varFilter, and required at least three variant reads as well as ≥20% variant reads for each called position, with Phred-like quality scores of at least 20 for single nucleotide variants (SNVs) and at least 50 for small insertions or deletions (indels). We used Annovar and custom scripts to annotate variants according to the type of mutation, occurrence in the Single Nucleotide Polymorphism database (dbSNP), Sorting Intolerant from Tolerant (SIFT) score, 1000 Genomes allele frequency, and National Heart, Lung and Blood Institute (NHLBI) exomes allele frequency.16 To identify potentially pathogenic variants we filtered out (1) synonymous variants or intronic variants other than those affecting the consensus splice sites; (2) variants seen in more than two of 416 exomes from patients with rare, monogenic diseases unrelated to JBTS that were sequenced at the McGill University and Genome Quebec Innovation Centre and (3) variants with a frequency greater than 0.5% in either the 1000 genomes or NHLBI exome datasets.
We first examined the eight exome datasets to look for rare variants in the 17 known JBTS genes (INPP5E[MIM613037], TMEM216[MIM613277], AHI1[MIM608894], NPHP1[MIM607100], CEP290[MIM610142], TMEM67[MIM609884], RPGRIP1L[MIM610937], ARL13B[MIM608922], CC2D2A[MIM612013], CXORF5[MIM300170], KIF7[MIM611254], TCTN1 [MIM609863], TCTN2[MIM613885], TMEM237[MIM614424], CEP41[MIM610523], TMEM138[MIM614459], C5ORF42[MIM614571],1–13 as well as in the JBTS candidate gene TTC21B(MIM612014).17 Five individuals from three families (II-1 from family 379, II-4, II-6 and IV-1 from family 385/447, and II-1 from family 492 online supplementary figure S2) were each found to carry two rare heterozygous variants in CC2D2A(NM_001080522.2). One in-frame amino acid deletion (c.3450_3452del[p.Val1151del]) and four different missense variants (c.3376G>A[p.Glu1126Lys], c.4559A>G[p.Asn1520Ser], c.4667A>T[p.Asp1556Val], c.4702T>C[p.Tyr1568His]) were identified, two of which, c.3376G>A(p.Glu1126Lys) and c.4667A>T(p.Asp1556Val), were identified previously in FC individuals with JBTS.12 The novel mutations c.4559A>G(p.Asn1520Ser) and c.4702T>C(p.Tyr1568His) are predicted to be damaging (by SIFT, Polyphen-2 and Mutation Taster) and neither variant has been reported in the Exome Variant Server (EVS; NHLBI GO Exome Sequencing Project), dbSNP135 or 1000 Genome datasets. These five CC2D2A mutations cluster in either the C2 domain (amino acids 1062–1174) or the C-terminal part of the protein, as do most missenses that cause JBTS.18 Segregation analysis revealed that all the affected individuals, but none of their unaffected relatives, were compound heterozygous for the mutations (see online supplementary figure S1). We conclude that these mutations are pathogenic and responsible for JBTS in these five individuals.
We also identified a frameshift mutation (c.8257_8258insA[p.Lys2753fs]) and a splice-site mutation (c.7400+1G>A) in C5ORF42(NM_023073.3) in individual II-2 from family 551. Sanger sequencing showed that the proband is compound heterozygous for these mutations. The splice site (c.7400+1G>A) mutation has been previously identified in patients with JBTS and shown to result in skipping of exon 35 and the creation of a premature stop codon while c.8257_8258insA(p.Lys2753fs), which is novel, is predicted to truncate C5ORF42 in the middle of its sequence, close to where other truncating mutations have been previously identified in JBTS patients.12 Both C5ORF42 mutations are thus considered pathogenic in this individual. Table 1 summarises the genotypes and phenotypes of these patients with mutations in CC2D2A and C5ORF42, as well as those of FC patients previously described with mutations in these genes. Individuals in our cohort with mutations in CC2D2A do not have any extraneural manifestations, and appear to have a milder phenotype, with all affected individuals walking independently before the age of 4 years, and intelligence ranging from normal to mild intellectual impairment. Individuals with mutations in C5ORF42 have a more variable phenotype. They have borderline to moderate cognitive impairment and variable age at walking, ranging between 30 months and 8 years. Some patients also showed limb abnormalities, including one individual with combined pre- and postaxial polydactyly, an unusual finding in JBTS, which is typically associated with postaxial polydactyly.
Genotypes and phenotypes of French Canadian individuals with JBTS
We then combined the exome data of the two remaining individuals with unexplained JBTS and the exome data of four individuals with unexplained JBTS from our previous study,12 making a total of six individuals from four different families. We analysed the data by looking for protein-coding genes that contained homozygous or multiple heterozygous variants in each affected individual. For multiplex families, we only considered genes with the same variants in the affected siblings (see online supplementary tables S1 and S2). Only one gene, TMEM231, harboured multiple rare mutations in more than one family. Three JBTS individuals from 2 families (II-1 and II-2 from family 387, and II-1 from family 483) harboured the same two variants in TMEM231(NM_001077418.1): c.12T>A(p.Tyr4*) and c.625G>A(p.Asp209Asn). Sanger sequencing showed that all affected individuals were compound heterozygous for these variants (figure 1A). The c.12T>A(p.Tyr4*) mutation targets exon 1 of the canonical isoform of TMEM231(NM_001077418.1; ENST00000258173), as well as the two other predicted protein-coding isoforms reported in the Ensemble Genome Browser. In ENST00000565067, it leads to the same nonsense change (p.Tyr4*), while in the longer isoform ENST00000398114, it abolishes the translation initiation methionine (c.2T>A[p.Met1?]), which would likely prevent translation of this isoform due to the absence of any other in-frame methionine in exons 1 and 2. The c.625G>A(p.Asp209Asn) causes the same amino acid change in the different TMEM231 predicted isoforms (figure 1C). It affects a highly conserved amino acid (figure 1D), and is predicted to be damaging by Polyphen-2 and Mutation Taster but not by SIFT. Both p.Tyr4* and p.Asp209N are extremely rare. Among the 416 in-house exomes, the c.12T>A(p.Tyr4*) was not found, and the c.625G>A(p.Asp209Asn) variant was seen in the heterozygous state in one FC individual. No additional TMEM231 coding/splicing variants were present in this individual's exome. To determine the carrier rate of c.625G>A and c.12T>A, we genotyped 385 healthy FC controls by Sanger sequencing, but did not find any carriers of either of these mutations, indicating that they are very rare. Only p.Asp209Asn is reported in the heterozygous state in the 1000 genomes and EVS, but at a very low frequency (0.01%), while p.Tyr4* is not reported in any of these public single nucleotide polymorphism (SNP) databases. Furthermore, no truncating mutations in TMEM231 were seen in 416 control exomes of patients with other rare diseases, and only one other truncating variant (stopgain SNV) is reported in EVS, at a frequency of 0.04%. For the three individuals with compound heterozygous TMEM231 mutations, we examined all SNV genotypes in regions surrounding the two mutations. This revealed a region of shared genotypes (two shared haplotypes) extending over at least 1.7 Mb, suggesting the existence of founder effects (see online supplementary table S3).

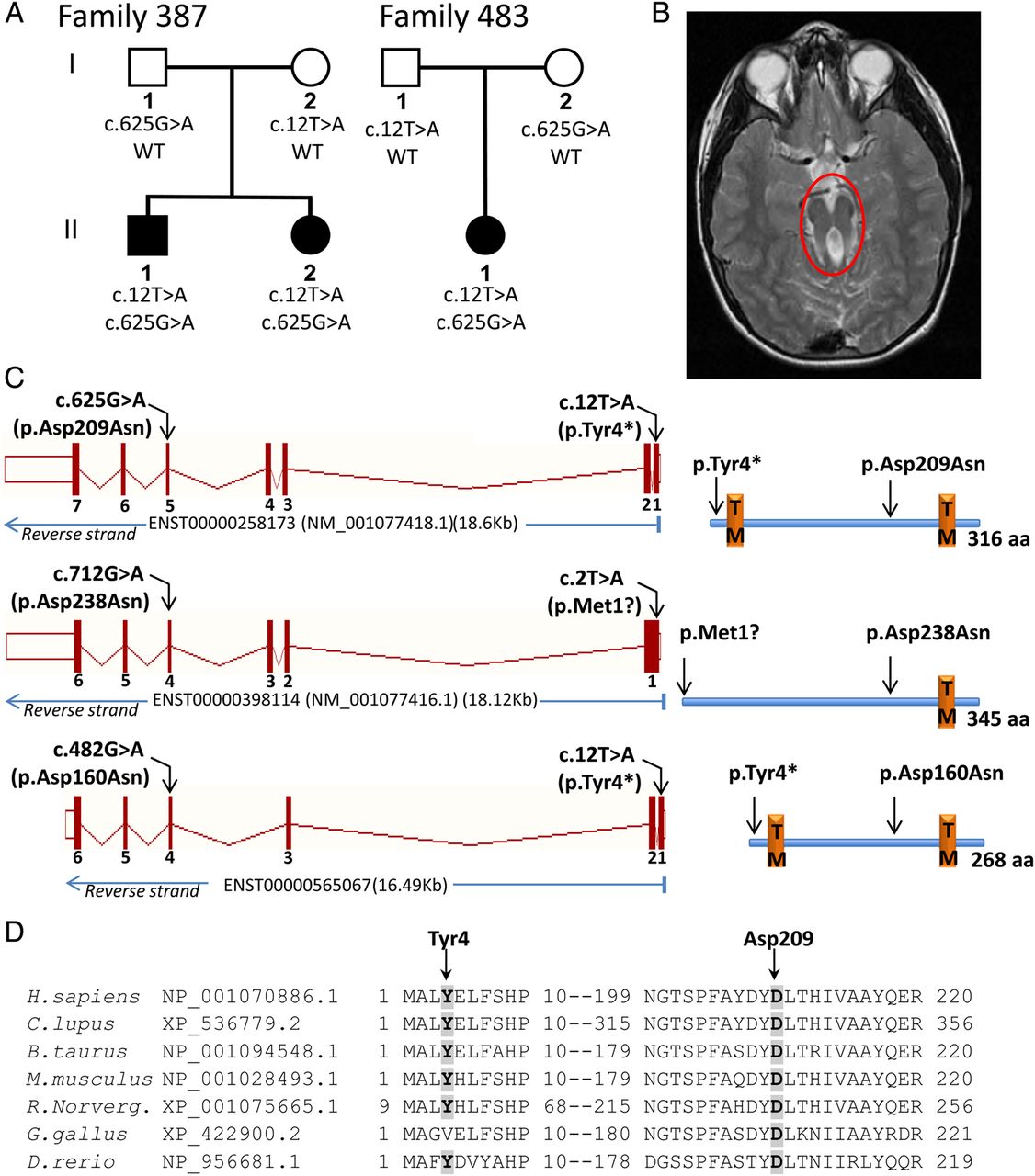{kind=link}
(A) Segregation of mutations in TMEM231 in JBTS families. (B) Brain MRI from individual II-1 from family 387 showing the ‘molar tooth sign’. (C) Left panel, scheme showing the presence of the mutations with respect to the different TMEM231 Ensemble-annotated transcripts predicted to produce proteins; right panel, the corresponding TMEM231 proteins are depicted in the right panel. TM, denotes the presence of a transmembrane domain, as predicted by SMART algorithm. (D) Amino acid conservation of the residues affected by the p.Tyr4* and p.Asp209Asn mutations in TMEM231. Amino acid alignments were generated using homologene (NCBI).
The three individuals with mutations in TMEM231 are among the most severely affected in our French–Canadian JBTS cohort. They are dependent in all activities of daily living, are non-verbal, and can take steps only if assisted with a walker. Both siblings from family 387 had significant aggressive and self-mutilating behaviour consisting of head banging and biting, requiring treatment with antipsychotic agents, mouth guard and protective helmet. Individuals II-2 from family 387 and II-1 from family 483 show extraneural manifestations consisting of retinopathy, bilateral macroscopic renal cysts (but normal renal function), and postaxial polysyndactyly of one foot (table 1 and see online supplementary figure S1, D-F).
The presence of rare and potentially deleterious mutations in TMEM231, which segregate with the disease in two unrelated FC families, strongly suggests that disruption of this gene causes JBTS in our subjects. The fact that the three individuals with the mutations in TMEM231 show a similar form of JBTS also supports the involvement of this gene. Furthermore, several observations indicate that TMEM231 plays a key role in the cilia, and physically interacts with known JBTS genes. TMEM231 encodes a transmembrane protein that localises at the base of the ciliary axoneme at the transition zone.19 Recently, TMEM231 was shown to be part of the B9 complex, which is required for a diffusion barrier between the cilia and plasma membranes that maintains the integrity of the cilia as a privileged membrane domain.19 The B9 complex includes at least 13 proteins (BD91, BD92, TCTN1, TCTN2, TCTN3, CC2D2A, TMEM216, TMEM67, TMEM237, TMEM231, MKS1, AHI1, TMEM17), all of which, with two exceptions (TCTN3, TMEM17), are involved in JBTS and/or Meckel–Gruber syndrome (MKS), a related ciliopathy.19 ,20 Proteomic studies using the MKS proteins BD91 or BD92 as baits have shown that TMEM231 is in a complex with TCTN1, TCTN2, MKS1, AHI1 and CC2D2A.19 siRNA knockdown of TMEM231 disrupts the integrity of the ciliary barrier and the localisation of components of the B9 complex at the transition zone, resulting in a reduction of cilia formation and ciliary localisation of signalling receptors.19 TMEM231 knockout mice die at E15.5 with severe vascular defects, and display typical features of ciliopathy, namely microphthalmia, polydactyly and abnormalities in patterning of ventral spinal cord.19 Altogether, these observations indicate that autosomal-recessive mutations in TMEM231 are a cause of JBTS.
JBTS in FCs show both locus and allelic heterogeneity. We identified three JBTS genes in this population with a total of 14 different alleles. Three mutations in C5ORF42, two mutations in CC2D2A and two mutations in TMEM231 were found in at least two unrelated affected individuals (table 1). Our analysis indicates that each of these mutations is located within a distinct haplotype in these individuals, suggesting the existence of multiple founder effects.12 Founder effects are typically associated with an increase in the frequency of a specific autosomal recessive allele, which is often accompanied by other alleles that remain at their usual background frequency. Interestingly, for each of these three JBTS genes, we found at least two founding mutations. It is likely that more of these complex founder effects will be unravelled with the use of genomic sequencing.
In summary, combining this study and our previous one, we were able to explain the underlying genetic cause of JBTS in 21/24 FC individuals using exome sequencing. In the course of this work, we identified TMEM231 as a novel JBTS gene. This discovery gives further support to the concept that JBTS results from disruption of the barrier at the ciliary transition zone.
Acknowledgments
Foremost, we thank the families who generously contributed their time and materials to this research study. This work was selected for study by the FORGE Canada Steering Committee, consisting of K Boycott (University Ottawa), J Friedman (University of British Columbia), J Michaud (Université de Montréal), F Bernier (University Calgary), M Brudno (University Toronto), B Fernandez (Memorial University), B Knoppers (McGill University), M Samuels (Université de Montréal), and S. Scherer (University of Toronto). We would like to thank Janet Marcadier (Clinical Coordinator) and Chandree Beaulieu (Project Manager) for their contribution to the infrastructure of the FORGE Canada Consortium. The authors wish to acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Génome Québec Innovation Centre, Montréal, Canada. JL Michaud is a National Scholar from the Fonds de la Recherche en Santé du Québec (FRSQ). M Srour holds a CIHR clinician-scientist training award.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors MS, FFH, JM, JLM: study design, data analysis and interpretation and manuscript writing and revision. JS: data analysis and manuscript writing and revision. GM, EL, LP, SD: laboratory follow-up of candidate variants and segregation studies. MS, JLM, LHO, MIS, VD, DA, EA, GS, BM: patient recruitment, examination and counselling. DL, GAR: contribution of control samples. CM: coordination of samples and patient consents.
-
Funding This work was funded by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research (CIHR) and the Ontario Genomics Institute (OGI-049).
-
Competing interests None.
-
Ethics approval Ethics Committee of Sainte Justine Research Center.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
WEB RESOURCES 1000 Genomes Project, http://browser.1000genomes.org/index.html dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/ Ensemble Genome Browser: http://www.ensembl.org ESP Exome Variant Serve (EVS)r: http://evs.gs.washington.edu/EVS/ Gene Ontology, http://www.geneontology.org/ Mutation Taster: http://www.mutationtaster.org/ NCBI homologene, http://www.ncbi.nlm.nih.gov/homologene NCBI Nucleotide database, http://www.ncbi.nlm.nih.gov/nuccore Online Mendelian Inheritance in Man (OMIM), http://www.omim.org Polyphen-2: http://genetics.bwh.harvard.edu/pph2/ SIFT: http://sift.jcvi.org/ SMART sequence analysis: http://smart.embl-heidelberg.de/