Article Text

Abstract
Background Constitutional DICER1 mutations were recently reported to cause familial pleuropulmonary blastoma (PPB).
Aim To investigate the contribution and phenotypic spectrum of constitutional and somatic DICER1 mutations to cancer.
Methods and results The authors sequenced DICER1 in constitutional DNA from 823 unrelated patients with a variety of tumours and in 781 cancer cell lines. Constitutional DICER1 mutations were identified in 19 families including 11/14 with PPB, 2/3 with cystic nephroma, 4/7 with ovarian Sertoli–Leydig-type tumours, 1/243 with Wilms tumour (this patient also had a Sertoli–Leydig tumour), 1/1 with intraocular medulloepithelioma (this patient also had PPB), 1/86 with medulloblastoma/infratentorial primitive neuroectodermal tumour, and 1/172 with germ cell tumour. The inheritance was investigated in 17 families. DICER1 mutations were identified in 25 relatives: 17 were unaffected, one mother had ovarian Sertoli–Leydig tumour, one half-sibling had cystic nephroma, and six relatives had non-toxic thyroid cysts/goitre. Analysis of eight tumours from DICER1 mutation-positive patients showed universal retention of the wild-type allele. DICER1 truncating mutations were identified in 4/781 cancer cell lines; all were in microsatellite unstable lines and therefore unlikely to be driver mutations.
Conclusion Constitutional DICER1 haploinsufficiency predisposes to a broad range of tumours, making a substantial contribution to PPB, cystic nephroma and ovarian Sertoli–Leydig tumours, but a smaller contribution to other tumours. Most mutation carriers are unaffected, indicating that tumour risk is modest. The authors define the clinical contexts in which DICER1 mutation testing should be considered, the associated tumour risks, and the implications for at-risk individuals. They have termed this condition ‘DICER1 syndrome’.
Accession numbers The cDNA Genbank accession number for the DICER1 sequence reported in this paper is NM_030621.2.
- DICER1
- microRNA
- cancer syndrome
- tumour predisposition
- genetics
- clinical genetics
- oncology
- paediatric oncology
Statistics from Altmetric.com
- DICER1
- microRNA
- cancer syndrome
- tumour predisposition
- genetics
- clinical genetics
- oncology
- paediatric oncology
Introduction
DICER1 is an RNase endonuclease essential in the production of microRNAs (miRNAs), which are non-protein-coding small RNAs that are estimated to regulate the expression of over 30% of protein-coding genes at the post-transcriptional level.1 2 miRNAs are transcribed as long precursors, known as pri-miRNAs, which are processed in the nucleus to produce pre-miRNAs.3 The pre-miRNAs are exported to the cytoplasm, where DICER1 processing generates a double-strand miRNA duplex.4 The duplex is unwound to generate the final miRNA, which interacts with mRNA to regulate gene expression, typically through translational repression or mRNA degradation.2 5 6
Over 900 human miRNAs are currently recognised.7 8 These have been implicated in a wide range of biological processes including metabolism, morphogenesis, cell fate determination, cell proliferation and apoptosis.9 10 There is increasing evidence implicating dysregulation of miRNAs in several human diseases, including cancer.11 12 Widespread alteration of miRNA levels is seen in cancers, and miRNA profiles characteristic of cancer type and stage are increasingly recognised.12 13 Furthermore, global downregulation of miRNAs due to abrogation of miRNA processing has been shown to promote tumorigenesis.14
Recently, germline-inactivating DICER1 mutations were shown to cause familial pleuropulmonary blastoma (PPB, OMIM 601200), a rare malignant lung tumour, primarily affecting children before age 6.15 16 By linkage analysis, Hill et al mapped a familial PPB gene to chromosome 14q. They considered DICER1 to be a promising candidate and identified pathogenic mutations in 11 families.15 This important finding raises a number of questions. First, what is the contribution of DICER1 mutations to non-familial, sporadic PPB? Mutations in some cancer-predisposition genes contribute appreciably to both familial and sporadic forms of disease, whereas for others the contribution to non-familial cases is small. Second, do constitutional DICER1 mutations predispose to tumours other than PPB? The International PPB Registry has collected information from over 200 PPB families, and a variety of different tumours have been reported in PPB cases and/or their relatives 17 18 However, it is unknown which tumour types are genuinely associated with PPB, which are related to DICER1 mutations, and which reflect ascertainment. Third, do somatic DICER1 mutations contribute to cancer? This is of particular interest as it has been proposed that somatic 14q loss, which has been reported in many cancers, may be targeted at DICER1.19–21
To address these questions, we have conducted exhaustive DICER1 sequencing, in >1600 patient samples, including constitutional DNAs from 823 individuals with a broad range of tumours, but particularly focusing on tumours that have been proposed to be associated with PPB (http://www.ppbregistry.org/pdf/Doc_D.pdf) (table 1). We also sequenced DICER1 in DNA from 781 cancer cell lines to assess the impact of somatic DICER1 mutation on cancer development (online supplementary table 1).
Tumours in individuals screened for constitutional DICER1 mutations
Methods
Samples
Constitutional DNA was extracted from EDTA venous blood samples and collected through the Factors Associated with Childhood Tumours (FACT) Study, the Royal Marsden Hospital cancer collections, and the Institute of Cancer Research UK-wide testicular germ cell tumour collections, all of which have been approved by an appropriate ethics board. All samples were obtained with full informed consent. The research was undertaken as part of the FACT Study, which was approved by the London Multicentre Research Ethics Committee (05/MRE02/17). The FACT Study aims to identify genetic factors that predispose to the development of childhood tumours (http://www.icr.ac.uk/fact). The National Registry of Childhood Tumours was used to identify the total number (indicated in parentheses) of cases nationally for PPB (20, five of whom were deceased), cystic nephroma (15, none deceased) and Sertoli–Leydig cell tumours (seven, none deceased) that had been registered since its inception in 1962. The clinicians were contacted to request that they recruit these patients to the FACT Study. In addition, a small minority of patients referred to our clinical genetics service with these tumours were recruited directly. Tumour DNA from DICER1 mutation-positive paraffin-embedded tissues was extracted using QIAamp DNA FFPE Tissue kit (Qiagen) according to the manufacturer's instructions. We analysed whole-genome amplified DNA from 781 cancer cell lines as part of the Cancer Genome Project, Cell Line Project (online supplementary table 1).
DICER1 sequencing
For analysis of the constitutional DNA, we designed PCR primers to amplify the 26 coding exons and intron–exon boundaries of DICER1 in a multiplex PCR (online supplementary table 2). Products were sequenced by capillary sequencing using the BigDye Terminator Cycle Sequencing Kit and an ABI3730 Genetic Analyser (Applied Biosystems, Foster City, California, USA). Sequences were analysed using Mutation Surveyor software V.3.20 (SoftGenetics). We only included samples in which at least 90% or more of the coding sequence was successfully screened in subsequent analyses. For the cancer cell lines, PCR primers that amplify 500 bp PCR products encompassing the 26 coding DICER1 exons and intron–exon boundaries were designed and sequenced as described above. Sequence traces were analysed using AutoCSA software,22 followed by manual inspection of putative variants. All putative variants were confirmed by bidirectional sequencing of a second independently amplified PCR product. Matched normal cell lines were available for 40 cell lines. The somatic status of variants identified in these 40 cell lines was determined by sequencing DNA from the corresponding normal. In the remaining cell lines, we assumed that cell line variants that were also identified in the constitutional DNA screen were not somatic. We evaluated the likely pathogenicity of sequence variants using Polyphen, SIFT and NNSplice software.
Results
Germline DICER1 mutation analysis
We identified pathogenic mutations in 19/823 index individuals (table 2 and online supplementary figure 1). Seventeen mutations led to premature protein truncation as a result of frameshift, nonsense or consensus splice-site mutations. Two mutations are missense alterations, for which there is substantial evidence of pathogenicity. First, they are the only two missense alterations in the 3214 chromosomes screened that are predicted to be pathogenic by SIFT and Polyphen. Second, both target highly conserved residues in the RNase III domain. Third, they are in the vicinity of a missense variant identified by Hill et al, which resulted in a similar DICER1 histochemical profile to truncating mutations.15 We also identified several non-pathogenic variants including nine missense variants, 23 synonymous variants, and five intronic variants (online supplementary table 3).
Probands and relatives with constitutional DICER1 mutations
Pleuropulmonary blastoma
We identified DICER1 mutations in 10 individuals with PPB and in the mother of a child that had died from PPB but from whom no sample was available. One child developed an intraocular medulloepithelioma 4 years after PPB. Three-generational pedigrees were available for most cases, and no relative had PPB. One sibling died from neuroblastoma; her DICER1 status is not known, but a mutation was present in her mother. There were three children with PPB, in whom we did not identify a DICER1 mutation.
Cystic nephroma
The most common reported association of PPB is cystic nephroma,17 a rare benign renal tumour that typically presents as a multicystic renal mass without solid nodules. It has a bimodal incidence with 50% occurring in children less than 4 years and 30% in the 5th and 6th decades.23 We had DNA from three unilateral childhood cases, and in two we identified truncating DICER1 mutations. One of the children had a half-sibling with cystic nephroma who also has the mutation. The child in the second DICER1 mutation-positive case was recently found to have a small lung cyst, which is being monitored but has not had histological evaluation.
Ovarian Sertoli–Leydig tumours
We analysed DNA from 30 individuals with sex cord tumours, of which six were ovarian Sertoli–Leydig tumours, which are sex cord tumours that exhibit testicular differentiation.24 The age range of diagnosis is 2–75 years, but ∼75% present in the second or third decades.24 We identified truncating DICER1 mutations in four individuals; three had young-onset bilateral ovarian Sertoli–Leydig tumours, and one had a unilateral ovarian sex cord tumour that could not be further classified because of necrosis. In one of the bilateral cases, Wilms tumour was previously present (see below). The mother of one patient had also developed a Sertoli–Leydig tumour at 21 years and carried the DICER1 mutation. She has subsequently had melanoma at 50 years, endometrial cancer at 62 years, and breast cancer at 68 years.
Wilms tumour
Wilms tumour is an embryonal cancer of the kidney that affects ∼1 in 10 000 children, usually before the age of 6 years.25 26 We analysed DNA from 243 patients with Wilms tumour. We identified one truncating DICER1 mutation, in a child who developed Wilms tumour of atypical histology at the unusually late age of 8 years. Four years after treatment the child developed bilateral ovarian Sertoli–Leydig cell tumours.
Medulloblastoma/infratentorial primitive neuroectodermal tumour (PNET)
Medulloblastoma is a PNET that arises in the posterior fossa.27 We analysed 84 childhood medulloblastoma/infratentorial PNET cases and identified one truncating mutation in a child of 13 years. No other information or samples were available.
Seminoma
We analysed DNA from 185 individuals with germ cell tumour, of which 128 had a family history of testicular cancer. We identified one missense DICER1 mutation, Q1580H. A maternal first cousin once removed of this proband developed testicular cancer at 27 years, but the proband's mother does not carry the DICER1 mutation. It is not possible to conclusively establish whether this mutation is pathogenic on the available evidence.
Intraocular medulloepithelioma
Intraocular medulloepithelioma, also known as dictyoma, is a very rare embryonal tumour, usually originating in the ciliary body of the eye, which most commonly occurs during childhood.28 One child with PPB also developed a dictyoma and has a DICER1 mutation.
Thyroid non-toxic goitres/cysts
In the PPB Registry, thyroid cancers and thyroid hyperplasia are reported in both probands and relatives of PPB cases. We analysed DNA from 88 patients with thyroid cancer, but did not identify any mutations. However, one proband and six relatives of DICER1 mutation-positive individuals developed thyroid cysts/multinodular colloid goitre between the ages of 9 and 30 years. All were non-toxic, associated with normal thyroid function, and non-malignant. Thyroidectomy was required in four patients because of recurrent disease.
Other tumours/cysts
We did not identify mutations in any of the other tumour types as detailed in table 1.
Family studies in DICER1 mutation-positive individuals
We had samples from both parents in 17 families in which a DICER1 mutation had been identified in an index individual. In each, the mutation had been inherited (table 2). We had grandparental samples for five families. In two families, the mutation was absent in the respective grandparents, indicating that the mutation had arisen de novo in the parent. In the other three families, the mutation was present in a grandparent.
We analysed DNA from 51 relatives, and we identified 25 relatives with DICER1 mutations. Of these, as described above, one mother had a Sertoli–Leydig tumour, one half-sibling had cystic nephroma, and six relatives had thyroid cysts/goitre. The remaining 17 individuals did not have clinical features or symptoms likely to be related to the DICER1 mutation, one relative having muscular dystrophy and another Wegener's granulomatosis.
Analysis of tumours from DICER1 mutation-positive individuals
We obtained eight tumours from six DICER1 mutation-positive individuals. This included three PPB, four Sertoli–Leydig tumours, and one cystic nephroma. We analysed each tumour for the relevant mutation. Each was heterozygous for the mutation—that is, the tumour showed a similar mutational profile to that in the blood and there was no loss of the wild-type allele in any tumour.
DICER1 mutation analysis in cancer cell lines
We screened 781 cancer cell lines (online supplementary table 1) for DICER1 mutations. These represent an extensive cross-section of cancers, but do not include many of the cancers in which we identified germline DICER1 mutations. Two hundred and six of the cell lines have previously been shown to have loss of heterozygosity of 14q encompassing DICER1.
We identified four truncating mutations in the 781 cell lines, and these were in microsatellite unstable lines. We identified 22 non-synonymous variants that were either proven to be somatic or in which normal DNA was not available for evaluation. None were predicted to be deleterious (online supplementary table 4).
Discussion
In 2009, germline DICER1 mutations were identified in familial PPB, adding to the list of rare familial cancer syndromes that have yielded critical evidence linking essential biological processes with cancer causation.15 In this study, we have expanded knowledge of the link between DICER1 and cancer. First, we demonstrate that germline DICER1 mutations are the major cause of PPB. In the UK, 20 cases of PPB have been registered in the National Registry of Childhood Tumours over the last 35 years. We were able to include 14 in this study, none of which had a family history of PPB. We identified DICER1 mutations in 11 of these cases. This represents one of the largest known contributions of germline mutations of a single gene to a specific tumour type. It is uncertain whether cryptic DICER1 mutations account for any of the negative cases, whether mutations in another gene can also cause PPB, or whether the remaining cases are not due to germline predisposition genes. However, it is clear that germline DICER1 mutations are the major cause of both familial and non-familial PPB.
We have also demonstrated that DICER1 mutations cause a range of phenotypes; not all families include PPB, and a high proportion of mutation carriers are clinically well. In view of this, we suggest that ‘DICER1 syndrome’ is a preferable term to ‘PPB familial tumour syndrome’, which has previously sometimes been used. The range of different tumours that can occur in individuals with DICER1 mutations is broad, and it is likely that more associated tumours will be identified as further mutation testing is undertaken. The contribution of DICER1 mutations to different tumours is very variable. The major tumours that occur in DICER1 syndrome appear to be PPB, cystic nephroma and ovarian Sertoli–Leydig tumour. Evidence for this comes from our mutational data, our clinical and mutational investigation of relatives of mutation-positive probands, and from the spectrum of tumours that have been documented in relatives of PPB cases in the PPB Registry.17 18 The contribution of DICER1 mutations to different tumours is also very variable. Although germline DICER1 mutations may contribute significantly to cystic nephroma, ovarian Sertoli–Leydig tumour and intraocular medulloepithelioma, they are unlikely to have a major impact on the incidence of Wilms tumour, medulloblastoma/infratentorial PNET or neuroblastoma.
In addition to tumours, DICER1 mutations also appear to confer a risk of thyroid cysts. One proband and six DICER1 mutation-positive relatives developed thyroid cysts in childhood or young adulthood. Although histology results were only available for two cases, both were non-toxic multinodular colloid goitres. This is of particular interest, as the gene for familial non-toxic multinodular thyroid goitre has been previously shown to localise to 14q.29
The mutation analyses in cancer cell lines suggest that somatic DICER1 mutations do not make a substantial contribution to cancer. This is in contrast with recent reports hypothesising that the 14q hemizygosity observed in 206/761 cell lines is targeted at DICER1.19–21 In the great majority of the cell lines, the 14q loss of heterozygosity extends over a very large area and includes many genes. If loss of DICER1 were the main driver, one would expect that somatic DICER1 mutations would occur in at least some of the cell lines with normal 14q chromosomes. However, we only identified four truncating mutations, and these were in microsatellite unstable lines and therefore unlikely to be driver mutations. Thus the somatic mutational profile of DICER1 appears to differ from that of other ubiquitously expressed, critically important genes, such as TP53 and RB1, which also predispose to rare childhood cancers when mutated in the germline.
The mechanism of DICER1 tumour predisposition also appears to differ from the majority of known cancer-predisposition genes and is likely to operate by a haploinsufficiency mechanism (figure 1). Our analysis of tumours showed no loss of the wild-type allele, and Hill et al showed retained DICER1 expression in tumour cells.15 Data from mice studies are also consistent with a haploinsufficiency model and indicate that, whereas monoallelic DICER1 inactivation promotes tumorigenesis, biallelic loss is inhibitory.20 21 Our data further suggest that, although inactivation of one DICER1 allele is the initiating event in DICER1 syndrome, presumably because it leads to dysregulation of miRNA levels, other events must be required for cancer to occur. It is not known what these additional events are, or how many are required for oncogenesis to proceed. However, the low frequency of tumours in DICER1 mutation carriers suggests that either more than one additional event is required and/or the likelihood of the event(s) occurring is small.

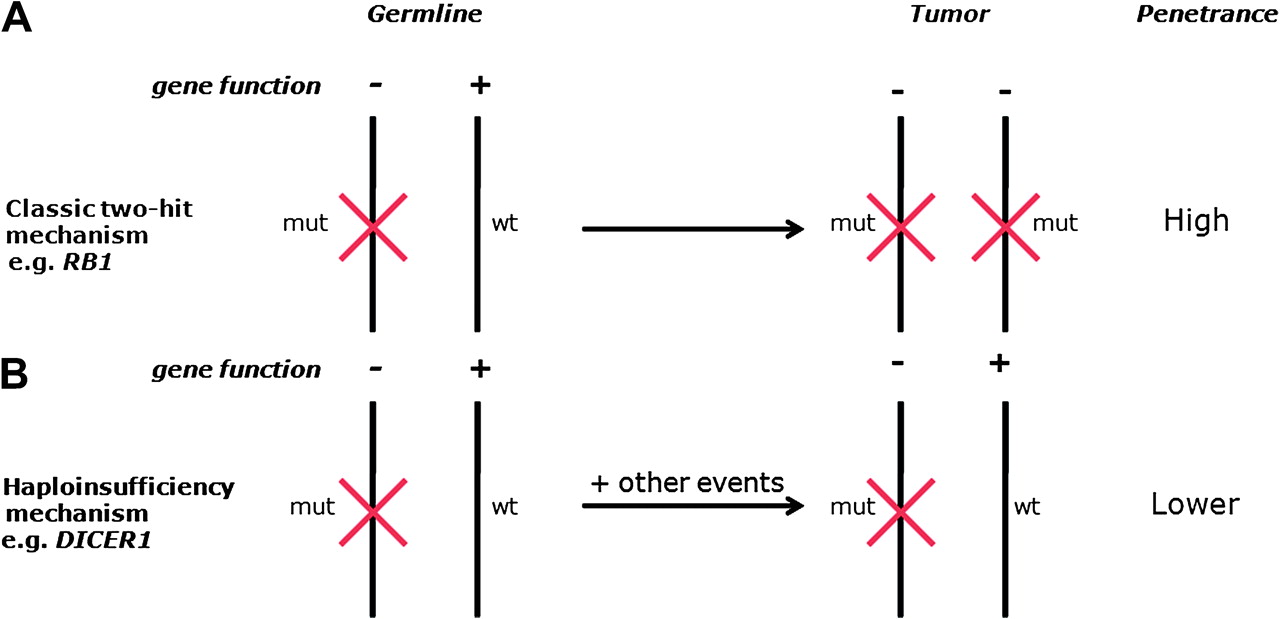{kind=link}
Different mechanisms of cancer predisposition resulting from germline mutations in tumour suppressor genes. (A) The classic two-hit mechanism, exemplified by retinoblastoma, involves a germline/constitutional mutation which constitutes the first hit and is present in every cell. A second mutation (hit) targeting the wild-type allele in a retinoblast has to occur for oncogenesis to proceed. (B) Haploinsufficiency mechanism, exemplified by DICER1 syndrome. A germline/constitutional mutation predisposes to tumours. One or more additional events are required for oncogenesis to proceed, but does not appear to include inactivation of the wild-type DICER1 allele. mut, mutant; wt, wild-type.
Our data demonstrate that the risk of tumours in DICER1 mutation carriers is low, and most mutation carriers do not develop tumours. This modest penetrance and the variable phenotype of DICER1 syndrome raise significant clinical challenges. We suggest that diagnostic DICER1 testing should be considered in individuals with possible PPB, cystic nephroma, ovarian Sertoli–Leydig tumour or medulloepithelioma. The prevalence of mutations in these conditions may be considerable, and identification of a DICER1 mutation can aid diagnosis and management, particularly for PPB, which can show significant clinical overlap with other types of lung cyst.18 30
The issue of surveillance in a pleiotropic condition of modest penetrance is also complex. To date, ad hoc, variable screening for PPB has been undertaken in individual families, often using lung CT, which can require an anaesthetic, involves radiation exposure, and is of unproven efficacy.31 Moreover, the natural history and appropriate management of such screen-detected lesions in a well child is unknown. In view of these considerations and the modest penetrance, we are currently operating an ‘open-door’ management policy with early investigation of potential tumour-related symptoms, but we are not undertaking routine surveillance in healthy mutation-positive individuals. This policy will be under continuous review, particularly over the next few years, when extensive expert discussions about the optimal management of DICER1 mutation carriers are likely to occur.
In this study, we clarify the phenotypes associated with constitutional DICER1 mutations and propose that the condition should be called ‘DICER1 syndrome’. In the future, additional research will hopefully further clarify the clinical features, tumour risks, and optimal management of this condition and will illuminate the mechanisms by which DICER1 haploinsufficiency predisposes to human disease.
Web resources
The URLs for data presented herein are as follows:
PPB Registry, http://www.ppbregistry.org/pdf/Doc_D.pdf
Cancer Genome Project, Cell Line Project, http://www.sanger.ac.uk/genetics/CGP/CellLines/
Polyphen, http://genetics.bwh.harvard.edu/pph/
Cancer Genome Project, Catalogue of Somatic Mutations in Cancer, http://www.sanger.ac.uk/perl/genetics/CGP/cosmic
Acknowledgments
We thank the children and families involved in the research, and the physicians, nurses and pathologists who referred families and provided samples. We are grateful for assistance with recruitment and discussions with the International PPB Registry. We thank Katrina Tatton-Brown, Helen Hanson, Trevor Cole, Anita Bayne, Margaret Warren-Perry, Darshna Dudakia, Polly Gibbs, Jessie Bull and Anna Zachariou for assistance with recruitment. We thank Katrina Spanova, Bernadette Ebbs and Deborah Hughes for running the ABI sequencers. We thank Ann Strydom for assistance with the manuscript. The research was carried out as part of the Factors Associated with Childhood Tumours (FACT) Study, which is a UK Children's Cancer and Leukaemia Group (CCLG) study.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Funding The Childhood Cancer Research Group receives funding from the Department of Health and the Scottish Ministers. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health and the Scottish Ministers. IS is supported by the Michael and Betty Kadoorie Cancer Genetics Research Programme. We acknowledge NHS funding to the NIHR Biomedical Research Centre. This work was supported by Cancer Research UK (grants C8620_A9024 and C8620_A8857) and the Institute of Cancer Research (UK).
Competing interests None.
Ethics approval This study was conducted with the approval of the NHS National Research Ethics Service.
Provenance and peer review Not commissioned; externally peer reviewed.