Article Text

Abstract
Background Nerve growth factor β (NGFβ) and tyrosine kinase receptor type A (TRKA) are a well studied neurotrophin/receptor duo involved in neuronal survival and differentiation. The only previously reported hereditary sensory neuropathy caused by an NGF mutation, c.661C>T (HSAN5), and the pathology caused by biallelic mutations in the TRKA gene (NTRK1) (HSAN4), share only some clinical features. A consanguineous Arab family, where five of the six children were completely unable to perceive pain, were mentally retarded, did not sweat, could not discriminate temperature, and had a chronic immunodeficiency, is reported here. The condition is linked to a new homozygous mutation in the NGF gene, c.[680C>A]+[681_682delGG].
Methods Genetic linkage and standard sequencing techniques were used to identify the causative gene. Using wild-type or mutant over-expression constructs transfected into PC12 and COS-7 cells, the cellular and molecular consequences of the mutations were investigated.
Results The mutant gene produced a precursor protein V232fs that was unable to differentiate PC12 cells. V232fs was not secreted from cells as mature NGFβ.
Conclusions Both the clinical and cellular data suggest that the c.[680C>A]+[681_682delGG] NGF mutation is a functional null. The HSAN5 phenotype is extended to encompass HSAN4-like characteristics. It is concluded that the HSAN4 and HSAN5 phenotypes are parts of a phenotypic spectrum caused by changes in the NGF/TRKA signalling pathway.
- Clinical genetics
- peripheral nerve disease
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Nerve growth factor (NGF) was discovered in the 1950s.1 It is translated as a precursor pro-protein, which undergoes successive N-terminal cleavage events to produce a biologically active C-terminal fragment called nerve growth factor β (NGFβ), which homodimerises and is secreted.2 The major NGFβ receptor is the neurotrophic tyrosine kinase receptor type A (TRKA) encoded by the gene NTRK1.3–5 The pan-neurotrophin receptor p75NTR is a secondary receptor for NGFβ.5 In mice, the NGF/TRKA pathway is essential for the differentiation and development of pain and temperature sensing neurons, particularly the C-fibres of peripheral nerves.6
The only reported family with a homozygous NGF mutation7–9 presented with congenital lack of pain appreciation, deficient temperature sensing and a lack of C-fibres, but normal sweating, immunity and cognitive abilities.7 This was described as HSAN5. Although NGFβ is the major TRKA ligand, many biallelic mutations in NTRK110 11 have been reported that cause more severe clinical findings. Patients with NTRK1 mutations, described as HSAN4, share the lack of pain appreciation and lack of C-fibres, but additionally present with anhidrosis and mild to moderate mental retardation, and recently an immune phenotype was also described.10 12 Our findings explain this apparent incongruence by extending the HSAN5 phenotype, caused by NGF mutations, to encompass clinical aspects previously linked only to NTRK1 mutations.
Methods
For details of all methods see supplemental data.
Clinical studies
The family was ascertained through a local clinical genetics service after they sought a diagnosis. Research ethics approval for this work was gained from the appropriate authorities in the United Arab Emirates and the UK. Nerve biopsy and formal assessments of pain were not considered justifiable.
Genotyping and mutation detection
Autozygosity mapping was performed on three affected family members. Data were analysed and concordant homozygous regions further investigated. Candidate genes were sequenced using patient genomic DNA.
Cell culture and transfection
Rat pheochromocytoma PC12 cells and Simian kidney COS-7 cells were kept in standard culture conditions and transiently transfected with either wild-type (WT) or mutant plasmid constructs.
Transfection plasmid construction
Full length NGF sequences were amplified from genomic DNA from a patient and a control, and cloned into pIRES2-AcGFP1. The missense mutation (c.661C>T) was generated using wild-type NGF-pIRES2-AcGFP1 as the template. FLAG-tagged NGF constructs were based on those previously described.13 All clones were confirmed by DNA sequencing.
PC12 differentiation assay
PC12 cells were grown on BD Matrigel coated glass coverslips and transfected with constructs expressing wild-type/mutant NGF, or empty vector (null control). As a positive control, 100 ng/ml murine submaxillary gland NGF was added to untransfected cells. Cells were methanol-fixed and mounted. Neurite length was compared between transfected cell populations.14 Experiments were conducted in duplicate and scored blind. Data are presented as mean ±SEM. The level of statistical significance for comparison was set at p<0.05.
ELISA and western blot analysis
PC12 and COS-7 cells were transfected with FLAG-tagged constructs. After 24–48 h, the media and cell lysates were collected. Total protein concentration was measured in duplicate. Enzyme linked immunosorbent assay (ELISA) for NGF was performed following the manufacturer's instructions. The level of statistical significance was set at p<0.05. For western blotting analysis, protein lysate and concentrated media extracts were electrophoresed on gradient gels, transferred to polyvinylidene fluoride (PVDF) membrane, blocked and incubated overnight with the relevant primary antibodies. Secondary antibody was added for 1 h. Signal was detected using ECL or ECL Plus (Amersham Biosciences, Little Chalfont, UK).
Results
Clinical studies
We ascertained a consanguineous Bedouin family with five affected children (figure 1A). Both parents and another child had a normal phenotype, without altered pain appreciation. The affected children were both male and female and ranged in age from 2–12 years. None appeared to experience pain. The first medical problems were of biting lips, tongue and digits without apparent discomfort (figure 1B, C). None could discriminate heat and cold, detect that spicy food was ‘hot’, nor did they ever sweat (anhidrosis). All had mild mental retardation evident by the age of 4 years. All were otherwise well grown and in good health, with a normal response to insect bites (which may serve as a proxy for an intradermal histamine flare test). Over time, all developed malar hypoplasia and sunken eyes, giving them a prematurely aged appearance, due to a loss of teeth from gingival disease—in three children all teeth were absent at the time of examination (figure 1C). All had suffered multiple, painless, injuries of varying severity (figure 1D). All had poor wound healing, usually requiring topical antibiotics. All had normal immunoglobulin values and white cell counts. More details are provided in the supplemental data.

Clinical phenotype and identification of the mutation. (A) Pedigree of the family showing relationship between affected and unaffected members. Clinical features of family members with NGF c.[680C>A]+[681_682delGG] mutation (CAdGG), showing damage to digits caused by biting (B), traumatic loss of tip of tongue and loss of teeth due to gingivitis(C), and a painless dislocation of the elbow joint (D). (E) Chromatograms comparing the wild-type carrier sequence (top) with that from an affected child (middle), and an alignment of the two sequences showing the altered base and two base pair deletion (bottom). (F) Alignment of part of the mature protein sequence showing the predicted altered amino acid sequence for the protein produced in the previously reported family (R221W) and in our family (V232fs) compared to wild-type NGF.
Genotyping and mutation detection
Mutation of NTRK1 was first considered, as the phenotype resembled HSAN4. However, the family was not linked to the locus, and affected individuals had different intragenic heterozygous single nucleotide polymorphism (SNP) haplotypes. Genome linkage using the eldest three affected individuals identified 11 concordant homozygous autosomal segments. Of these, only two contained genes implicated in nociception: GTP cyclohydrolase (GCH1) on chromosome 14q22, and NGF on chromosome 1p13.15
Sequencing of NGF revealed a homozygous mutation, c.[680C>A]+[681_682delGG] (hereafter referred to as CAdGG) (figure 1E), which segregated faithfully within the family, was not present in 320 ethnically matched control chromosomes or reported in human genomic databases. The mutation occurs in the single translated exon of NGF and leads to the bases ‘CGG’ being changed to ‘A’. The resultant frame-shift is predicted to replace the terminal 15 amino acids with a novel 43 amino acid terminal sequence (V232fs) (figure 1F). Two evolutionarily invariant cysteine residues at positions 229 and 231 are preserved, which are involved in disulfide bond formation in wild-type NGF.16 The CAdGG mutation creates additional cysteine residues in the novel carboxy terminus with two, C241 and C243, potentially able to compete in disulfide bond formation (figure 2B).

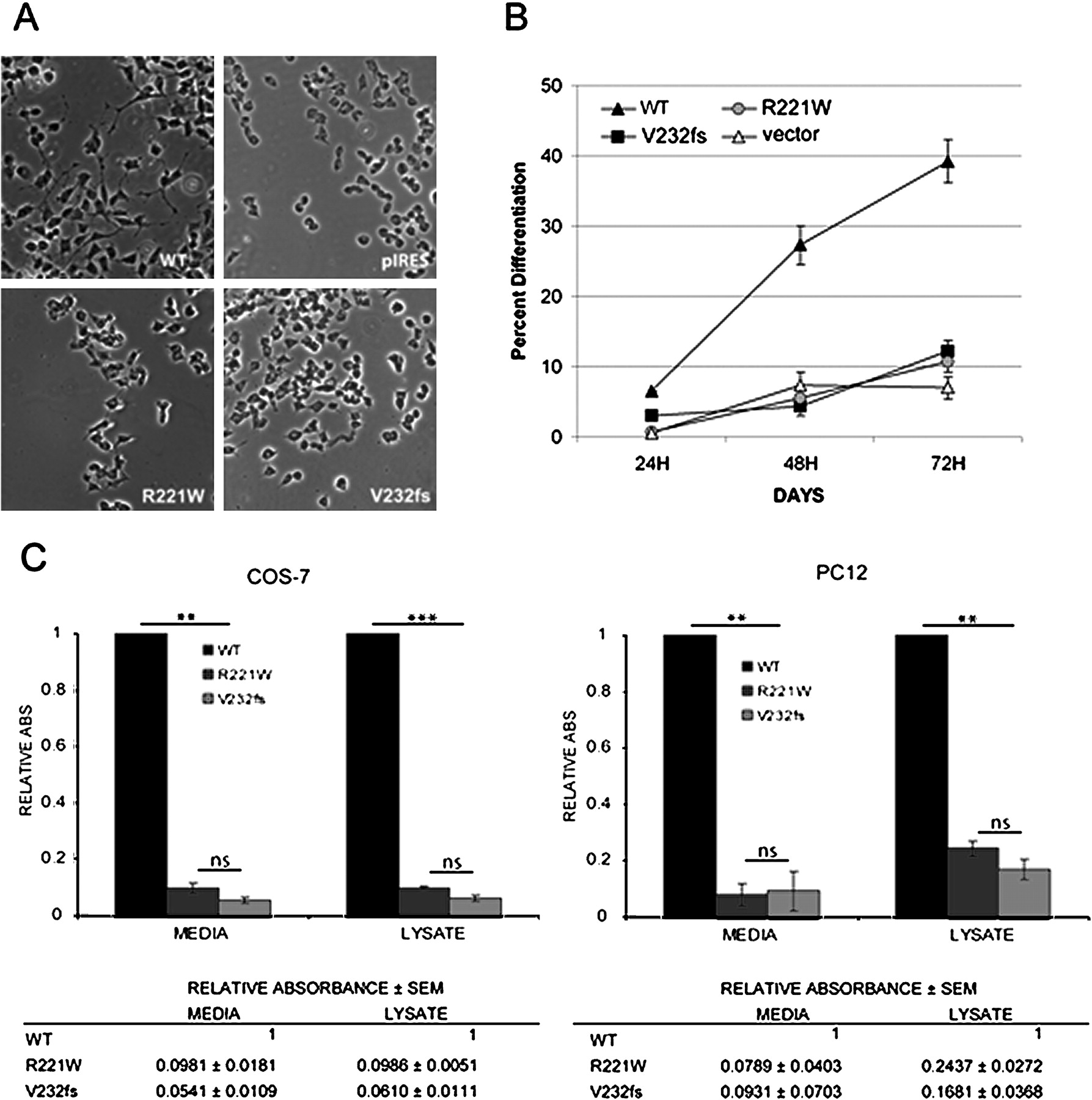
V232fs and R221W fail to induce PC12 differentiation and are not secreted from the cell. (A) PC12 cells transfected with empty vector (pIRES) or vector containing wild-type (WT), CT (R221W) or CAdGG (V232fs) mutant NGF genes. (B) Cells were monitored over a 72 h time course, and the percentage of differentiated cells was assessed at 24 h intervals. Images showing the relative differentiation of cells expressing WT (top left), R221W (bottom left), V232fs (bottom right) proteins, and empty vector control (top right) are shown. Amplification ×100. (C) Graph showing the percentage of differentiated cells for each of the proteins under study over 3 days. Two-way analysis of variance (ANOVA) analysis found significant differences between WT and both mutants at all the time points (p<0.01). There was no consistent difference between both mutants. Error bars represent mean ±SEM. COS-7 cells were transfected with empty vector or a vector containing wild-type, or either of the mutant (CT and CAdGG) NGF genes. 48 h after transfection, the media from the cells expressing WT, R221W or V232fs NGF was collected and concentrated, and cells were lysed. Total protein concentration was measured for each sample, before being assayed for NGF by ELISA. Results shown are the average of two independent experiments. Analysis with a one-way repeated measures ANOVA found NGF levels in wild-type were significantly higher than in either mutant for both the cellular extracts and media (p<0.01).
Analysis of a cohort of six consanguineous and 30 non-consanguineous individuals with an HSAN4- or HSAN5-like phenotype did not identify further NGF mutations, reaffirming that it is a rare cause of hereditary neuropathy.
We hypothesised that V232fs would have a significantly altered protein structure which could affect protein function. The previously reported NGF mutation, c.661C>T (referred hereafter as CT), was missense and led to the alteration of the invariant amino acid arginine 221 to tryptophan (R221W).
PC12 differentiation studies
To investigate the pathogenicity of our mutation we created constructs containing wild-type NGF, CT, or our CAdGG mutation.
We first asked whether either mutant could activate the TRKA receptor. For this, we took advantage of the characteristics of PC12 cells, which normally divide in culture in an undifferentiated state, but undergo mitotic arrest and neural differentiation upon NGF exposure (figure 2A).17 By comparing the percentage of differentiated cells induced by expression of wild-type, R221W or V232fs NGF, compared with an empty vector transfected control, we observed that unlike wild type NGF, both mutant proteins failed to effectively induce PC12 differentiation over 3 days (figure 2B). The difference in differentiation between both mutants and wild-type was significant on all days (p<0.01), but not between the two mutants (figure 2B). Therefore, both mutants were essentially unable to activate the TRKA receptor.
Analysis of intracellular processing of pro-NGF and NGFβ secretion
We looked next for evidence of pro-NGF and NGFβ secretion. Media and cell lysates from PC12 and COS-7 cells transfected with constructs containing FLAG-tagged wild-type, CT and CAdGG NGF genes or the empty vector (pcDNA) were assayed for the amount of NGF and NGFβ present by ELISA. The NGF signals from each sample were normalised as a function of the wild-type NGFβ signal. For both cell lines, a significant difference in secretion is seen between WT NGF and either mutant protein (p<0.001), but not between the two mutant proteins (figure 2C). However, although very sensitive, the ELISA cannot distinguish between failed processing and defective secretion. As an ELISA detects native proteins, it is also possible that the mutant proteins were not efficiently recognised, particularly in the case of V232fs, with its altered C-terminus. Therefore denaturing SDS PAGE was used to examine exactly which protein forms were present in all samples.
Western blotting analysis of cell lysates and media from transfected COS-7 cells confirmed that wild-type NGF was processed and secreted efficiently. In contrast, both mutants showed higher levels of pro-NGF (confirmed by detecting the FLAG epitope tag) in the cell lysates compared to wild-type NGF. In addition, while a very small amount of mature R221W NGF could be detected in the media, there was no detectable release of mature V232fs NGF above background (figure 3). Equal total protein concentrations were loaded, and even loading confirmed using detection of β-Actin. Similar results were seen in PC12 media extracts, although release of R221W was no longer detectable in the media from PC12 cells, most probably due to the lower transfection efficiency in this cell line (see supplemental data).

{kind=link}
{kind=link}
{kind=link}
V232fs is not processed while R221W is processed poorly. To determine whether the levels detected by ELISA represented precursor NGF or mature NGFβ, extracts were subjected to SDS PAGE, transferred to PVDF and the membranes probed with anti-NGF antibody to detect both precursor and mature forms (top), anti-FLAG specific for precursor form only (middle), and β-actin as a loading control (bottom). Both mutants seemed to fail to complete processing, although a faint band can be detected for the CT mutant in the media extract.
These results suggest that both NGF mutations impair processing of the pro-protein. Immunocytochemistry of transfected COS-7 and PC12 cells to identify the subcellular localisation of R221W and V232fs found no clear difference in localisation pattern between mutants and the wild-type protein (see supplemental data).
Discussion
We report a family where five affected children have a congenital inability to feel pain, anhidrosis, defective temperature sensing, mild mental retardation, and an immune deficiency. The parents and a sixth child have no clinical phenotype and in particular do not report a high pain threshold, nor have they suffered any painless injuries or particular medical problems. This family has a novel homozygous NGF mutation c.[680C>A]+[681_682delGG].
The family that define the HSAN5 phenotype has a homozygous NGF mutation c.[661C>T] resulting in a congenital inability to feel pain and defective temperature sensing, but conspicuously lacking the anhidrosis, mild mental retardation, and slow wound healing seen in our family.
As TRKA is the major NGF receptor, the clinical differences between HSAN4, caused by NTRK1 mutations, and HSAN5, were puzzling. Based on our findings, we suggest that the c.[661C>T] mutation is hypomorphic and retains some residual activity, while most NTRK1 mutations and our NGF mutation c.[680C>A]+[681_682delGG] are functionless.18 We suggest HSAN4 and HSAN5 form a phenotypic spectrum caused by deficiencies in the NGFB/TRKA pathway. Our data support this, as does the report of a child with a homozygous NTRK1 mutation and HSAN5-like symptoms.19 Furthermore, HSAN4 and HSAN5 have identical peripheral nerve biopsy findings. Clinically, the HSAN4 and HSAN5 phenotypes suggest that C-fibres are essential to pain sensing.
We assessed the functional significance of the CAdGG NGF mutation and compared it to the previously reported mutation, CT.13 We found that our mutation failed to differentiate PC12 cells. Within experimental constraints we detected no consistent difference between the two mutations. Our initial hypothesis was that the CAdGG mutation would significantly alter the protein tertiary structure and affect dimerisation.16 20 Instead we found that the V232fs NGF protein was not processed, while R221W NGF was processed very poorly (in agreement with the previous study13). Analysis showed that a small amount of R221W NGF was secreted into the media. In contrast, V232fs NGF was undetectable by either western blot (denatured) or ELISA (native peptide).
One aspect that seems to contradict a more severe effect for CAdGG is the absence of phenotype in the parents, both carriers of a mutated copy of the gene. In the Swedish family there is evidence of variable neuropathic symptoms among the carriers.21 We hypothesise that the R221W protein may have a small dominant-negative effect in Swedish family heterozygotes, as low levels of secreted R221W may interfere with wild-type NGFβ homodimerisation, resulting in a partially penetrant phenotype. In our family, we expect the carriers to effectively express only wild-type NGF, since V232fs is not secreted. Therefore, the absence of phenotype in the parents of our patients further supports, and indirectly validates, our conclusion that CAdGG is a null mutation.
We conclude that HSAN4 and HSAN5 represent a clinical spectrum with all cases sharing defective pain and temperature sensing sensation, while anhidrosis, mental retardation, and immune deficiency are present in more severe cases. Therefore both genes should be investigated in families presenting any set of symptoms associated with both neuropathies.
Web resources
Online Mendelian Inheritance in Man (OMIM) at http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim&itool=toolbar
Human Genome Browser at http://www.genome.ucsc.edu/
Primer3 for sequencing primer design at http://www.frodo.wi.mit.edu/primer3/
Acknowledgments
We thank the families and their physicians for their help. Thanks to Frank Reimann and Fiona Gribble for expert technical advice and support. We gratefully acknowledge funding for OPC and GKT from the Wellcome Trust, AKN from Pfizer (UK), and JJC from Action Medical Research.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
OPC and GKT contributed equally to this work.
Funding Action Medical Research, UK; Wellcome Trust; Pfizer, Sandwich, UK.
Competing interests None
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the relevant authorities in the United Arab Emirates and the UK (Cambridge Research Ethics Committee 05/Q0108/402).
Provenance and peer review Not commissioned; externally peer reviewed.