Article Text

Abstract
Background Leigh syndrome is an early onset, progressive, neurodegenerative disorder with developmental and motor skills regression. Characteristic magnetic resonance imaging abnormalities consist of focal bilateral lesions in the basal ganglia and/or the brainstem. The main cause is a deficiency in oxidative phosphorylation due to mutations in an mtDNA or nuclear oxidative phosphorylation gene.
Methods and results A consanguineous Moroccan family with Leigh syndrome comprise 11 children, three of which are affected. Marker analysis revealed a homozygous region of 11.5 Mb on chromosome 20, containing 111 genes. Eight possible mitochondrial candidate genes were sequenced. Patients were homozygous for an unclassified variant (p.P193L) in the cardiolipin synthase gene (CRLS1). As this variant was present in 20% of a Moroccan control population and enzyme activity was only reduced to 50%, this could not explain the rare clinical phenotype in our family. Patients were also homozygous for an amino acid substitution (p.L159F) in C20orf7, a new complex I assembly factor. Parents were heterozygous and unaffected sibs heterozygous or homozygous wild type. The mutation affects the predicted S-adenosylmethionine (SAM) dependent methyltransferase domain of C20orf7, possibly involved in methylation of NDUFB3 during the assembly process. Blue native gel electrophoresis showed an altered complex I assembly with only 30–40% of mature complex I present in patients and 70–90% in carriers.
Conclusions A new cause of Leigh syndrome can be a defect in early complex I assembly due to C20orf7 mutations.
- C20orf7
- Leigh
- complex I
- OXPHOS
- genetics
- clinical genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Leigh syndrome (MIM 256000) is an early onset, progressive, neurodegenerative disorder with a characteristic neuropathology consisting of focal, bilateral lesions in one or more areas of the central nervous system, including basal ganglia, thalamus, cerebellum, brainstem and spinal cord. The lesions are areas of demyelination, gliosis, necrosis, spongiosis, or capillary proliferation. Clinical symptoms depend on which areas of the central nervous system are affected. The most common underlying cause is a defect in oxidative phosphorylation (OXPHOS).1 Leigh syndrome can be associated with a deficiency of any of the mitochondrial respiratory chain complexes.2 The disorder is genetically heterogeneous and can be caused by mutations in the mtDNA and a variety of nuclear genes of the OXPHOS and pyruvate dehydrogenase system.
In structural complex I genes, mutations have been found in the mtDNA ND1,3 ND2,4 ND3,5 ND5,6 and ND67 genes and in the nuclear NDUFV1,8 NDUFS1,9 NDUFS3,10 NDUFS4,11 NDUFS7,12 NDUFS8,13 NDUFA1114 and NDUFA215 genes. In addition, mutations have been reported in the genes for the flavoprotein subunit A of complex II (SDHA16) and for the complex III assembly factor BCS1L.17 Mutations in complex IV genes include mtDNA encoded MTCO318 and nuclear encoded COX10,19 COX15,20 SCO2,21 and SURF1,22 which are involved in complex IV assembly. Several mutations were detected in the mtDNA encoded ATPase 6 (complex V).23 Additional mtDNA mutations have been described for the tRNA genes for valine,24 lysine,25 tryptophan26 and leucine.27
Leigh syndrome may also be caused by mutations in components of the pyruvate dehydrogenase complex (PDHA1 pyruvate dehydrogenase, α1-subunit and DLD, dihydrolipoamide dehydrogenase)28 29 and in the gene encoding the leucine-rich PPR motif containing protein (LRPPRC).30 Due to this extreme genetic heterogeneity, in many cases it is difficult to establish a genetic diagnosis in Leigh syndrome, especially if no additional clinical or biochemical data are available to pinpoint specific candidate genes. In order to identify the genetic cause in a consanguineous Moroccan family we performed homozygosity mapping and positional candidate gene analysis. A pathogenic amino acid substitution was identified in a new complex I assembly factor C20orf731 which caused Leigh syndrome with a diminished complex I activity, making defective complex I assembly an important pathogenic cause of Leigh syndrome.
Subjects and methods
Subjects
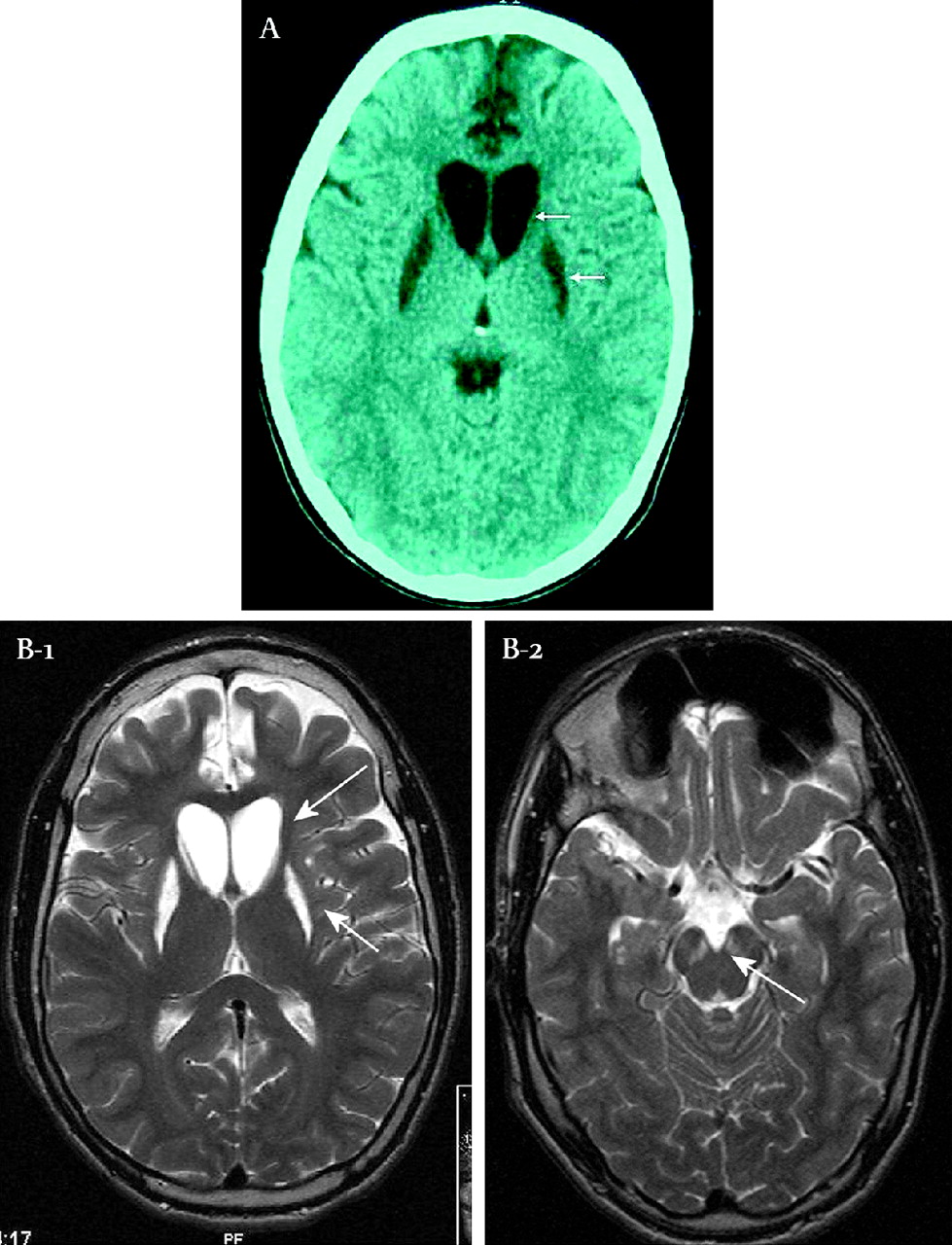
In a consanguineous family from Morocco with 11 children, three boys presented with Leigh syndrome (figure 1), two of whom were clinically investigated. The third affected sib succumbed for unknown reason at the age of 36 years in Morocco. The disease course is comparable in the two living patients (age 29 and 23 years). At the age of 3 they presented with a progressive spasticity with involvement of arms and legs. At the age of 5 they developed a diminished facial expression. Computed tomography (CT) scanning of the brain of patient IV7 at the age of 5 years showed hypodensity and slight atrophy of the caudate nuclei and the putamen (figure 2a). There is a widening of the frontal horns of the cerebral ventricles. One year later an extrapyramidal movement disorder with dystonic posturing of the right foot was noted. In the years following, the dystonia evolved in a more choreadystonic movement disorder. A delay in mental development became apparent. The dystonia hampered swallowing, necessitating nasal tube feeding. No signs of sensory nerve conduction loss were observed. At the age of 18 years both patients were moderately retarded with an IQ of about 50, had a remarkable good temper, and were severely handicapped by the dysarthria, the dystonic posturing, and the spastic tetraplegia. Their length was just below the third centile, but this was not different from the unaffected sibs and parents.

Pedigree of a consanguineous Moroccan family with three patients with Leigh syndrome. Black symbols indicate affected, white symbols unaffected subjects. The genotypes for the c.477A→C mutation in exon 5 of the C20orf7 gene are indicated below the tested individuals. The proband is indicated by an arrow.

Computed tomography (CT) and magnetic resonance imaging (MRI) of patients IV7 and IV11. respectively. (A) CT scan of patients IV7 brain at the age of 5 years. The CT scan shows a hypodensity and slight atrophy of the caudate nuclei and the putamen (arrows) and a widening of the frontal horns of the cerebral ventricles. (B-1) Cranial MRI of patient IV11 at the age of 23 years. Prolongation of T2 weighted signals in the residual part of the nucleus caudatus and putamen and (B-2) at the level of the midbrain of the substantia nigra (arrow).
In both patients the electroencephalogram (EEG) was normal. Pattern visual evoked responses demonstrated normal responses with N135 at 141 ms. Cranial magnetic resonance imaging (MRI) of patient IV11 at the age of 23 years showed a small residual nucleus caudatus and lesions in the basal ganglia consisting of prolongation of both T1 and T2 weighted signals in the caudate nucleus, putamen, the substantia nigra and a discrete abnormality in the peri-aquaductal grey area, and also discrete bifrontal global atrophy (figure 2b). After reaching puberty, the progression of the disease seemed to stop or slowed down. Orthopaedic interventions were necessary, because of complications of the movement disorders such as development of an equinovarus contracture in both feet and the development of a c-shaped scoliosis. Oral daily drug treatment consisted of carnitine 330 mg, riboflavin 30 mg, biotin 10 mg, and thiamine 100 mg. Parents and the other children are in good health and further family history is unremarkable. Informed consent was provided by the family for scientific investigations and publication.
Metabolic and enzymatic measurements
Routine metabolic workup was performed on the blood, urine and cerebrospinal fluid of the patients to rule out other inborn errors of metabolism including blood glucose, lactate, amino acids, acid/base status, ammonia, creatine kinase, carnitine, acylcarnitines, very long chain fatty acids, uric acid, B12, folate, cholesterol, isoelectric focusing of transferrin (for CDG, congenital disorders of glycosylation), biotinidase, lysosomal enzymes, purine and pyrimidine values; urine amino acids, organic acids, oligosaccharides, and mucopolysaccharide screening; cerebrospinal fluid (CSF) glucose, lactate, amino acids, and neurotransmitter measurements. A needle muscle biopsy from vastus lateralis muscle was performed at 22 and 29 years of age in patients IV11 and IV7, respectively. Complex I activity was determined in duplicate in these biopsy specimens and in peripheral blood lymphocytes (PBMCs) of patients IV7 and IV11 and their unaffected brother IV10. Assays to determine complex I and citrate synthase activities and protein content of the mitochondrial fractions were performed as described before.32–35
Fibroblasts were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco by Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 50 μg/ml of uridine (Acros Organics Geel, Belgium) and 50 units/ml penicillin and 50 μg/ml streptomycin (BioWhittaker, Walkersville, MD, USA). To isolate mitochondria, cells were resuspended in isolation buffer containing 0.25 M sucrose, 10 mM Tris-HCl pH 7.5, and 1 mM EDTA and homogenised on ice (10 strokes at 1500 rpm). Homogenates were centrifuged at 1600 g at 4°C for 10 min to remove cell debris and nuclei. Subsequently, mitochondria were pelleted from the supernatant at 10 000 g at 4°C for 10 min. Cardiolipin synthase (CLS) activity was determined in mitochondrial membranes after swelling the isolated organelles twice in 10 mM Bis-Tris propane-HCl buffer pH 7.4 and sedimenting mitochondrial membranes, which were finally resuspended in this Bis-Tris propane-HCl buffer containing 50% glycerol to a protein concentration of about 1 mg protein/ml. Mitochondrial protein, 1–6 μg, was used for CLS assays in a total volume of 50 μl, as described before36 except that the reaction mixtures were incubated at 37°C for 1 h.
Homozygosity mapping
Homozygosity mapping was performed, using the Affymetrix GeneChip Human Mapping 10K 2.0 Array (Santa Clara, CA, USA) for a whole genome analysis. Samples were processed and labelled according to the instructions of the manufacturer, hybridised in a GeneChip hybridisation oven followed by wash and stain with the GeneChip Fluidics Station 450, and scanning with the GeneChip Scanner 3000 (Affymetrix). Genotypes were generated by the GeneChip DNA analysis software (GDAS). The Copy Number Analysis Tool (CNAT, Affymetrix) was used to detect homozygosity regions in patient samples. Candidate regions were defined as homozygosity regions present in the patient samples but not in other family samples. Parametric lod scores were calculated using the Merlin package (version 1.1.2)37 with a recessive disease model.
Mutation analysis
The mtDNA was screened for deletions by long range polymerase chain reaction (PCR) and heteroplasmic point mutations by denaturing high performance liquid chromatography (DHPLC) analysis as described before.38 The exons and flanking introns of the human CRLS1 and C20orf7 genes were amplified with specific intronic primers (supplementary data table 1).
PCR was performed with 50 ng DNA using Taq-polymerase and buffer (Invitrogen, Carlsbad, CA, USA) with a final concentration of 1.5 mM MgCl2. Cycle conditions were: 94°C for 5 min, followed by 33 cycles of 94°C for 1 min, 54°C for 1 min, and 72°C for 1.5 min with a final elongation step of 72°C for 7 min. PCR products were directly sequenced with the PRISM Ready Reaction Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on an ABI3100 automatic sequencer (Applied Biosystems).
Mutation specific restriction digestion
A mutation specific restriction digestion assay was developed for the c.477A→C substitution in the C20orf7 gene. A total of 10 μl PCR product of exon 5 was digested for 1 h at 37°C with NheI and MseI, followed by a heat inactivation for 20 min at 65°C. MseI does not cleave if the mutation is present. Nhe1 was added to eliminate a constant band of 127 bp, which would otherwise trouble the interpretation. After digestion, the samples were resolved on a 3% agarose gel presenting a fragment of 109 bp for wild type samples compared to a 130 bp fragment for the mutant.
Blue native-polyacrylamide gel electrophoresis (BN-PAGE)
Mitochondria were isolated from PBMCs33 34 and incubated for 10 min on ice in lysis buffer (50 mM NaCl, 5 mM aminocaproic acid, 50 mM imidazole, pH 7.0) containing 1% Triton X-100 and supplemented with complete protease inhibitors (Roche Diagnostics GmbH, Mannheim, Germany), 1 mM 4-(2-aminoethyl)-benzenesulphonyl-fluoride hydrochloride (Roche) and 2 mM diisopropyl fluorophosphate (Fluka Chemica, Sigma-Aldrich, St. Louis, MO, USA). Next, 2.5 μg of PBMC mitochondrial protein was processed for one dimensional BN-PAGE as previously described39 using NativePAGE 3–12% gels (Invitrogen, Breda, The Netherlands) and Western transferred on PVDF membrane. Immunoreactive proteins were detected by mouse monoclonal antibodies against complex I subunits ND1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), NDUFA9 and complex II subunit 70 kDa Fp (MitoSciences, Eugene, OR, USA). Proteins were visualised using biotinylated secondary antibodies against mouse IgG (Amersham Biosciences, Little Chalfont, Bucks, UK), streptavidin-biotinylated horseradish peroxidase complex (Amersham) and SuperSignal West Femto (Pierce Biotechnology, Rockford, IL, USA). The molecular weights of the protein bands were estimated by a NativeMark Unstained Protein Standard (Invitrogen). Images were acquired using a calibrated densitometer (GS-800; Bio-Rad, Bio-Rad Laboratories, Hercules, CA, USA) and quantified using PDQuest Advanced software package (version 8.0.1 build 055; Bio-Rad).
Results
Metabolic and enzymological studies of two sibs with Leigh syndrome
Routine metabolic workup in urine and blood plasma of patients IV7 and IV11 only revealed a slight increase in blood alanine values without a raise in blood or urine lactic acid. Glucose tolerance test was normal. Also the methionine, vitamin B12, folic acid, purines and pyrimidines, and S-adenosylmethionine (SAM) concentrations were in the normal range (86–128 nmol/l). Cerebrospinal fluid examination at the age of 5 years of patient IV7 showed an increased lactic acid of 5 mmol/l (controls<2.0 mmol/l). Complex I activity, normalised by citrate synthase activity, was in muscle and PBMCs of patient IV7 respectively 36% and 6% of the controls and in patient IV11 respectively 48% and 33%. Activities of other OXPHOS complexes were only available for patient IV11 and were 105%, 123% and 87% of the control values for complex II, III and IV, respectively. Morphological studies show an increase in average fibre diameter. Staining for succinate tetrazolium reductase and NADH tetrazolium reductase showed increased subsarcolemmal activities. Staining for cytochrome c oxidase demonstrated some COX negative fibres.
Homozygosity mapping in the Leigh syndrome family
Molecular genetic analysis was initially directed at the mtDNA. First, the entire mtDNA of the muscle sample of patient IV7 was tested for deletions by long range PCR, but no mtDNA deletion was observed (data not shown). Next, the mtDNA was screened by DHPLC analysis,38 but no heteroplasmic point mutations were detected, making the involvement of a nuclear gene likely. Both parents, two affected (IV7 an IV11) and four unaffected sibs (IV1, IV5, IV6 and IV10) were tested with the Human Mapping 10K GeneChips. CNAT analysis revealed a large homozygosity region of 11.5 Mb on chromosome 20 (from 2.4 Mb to 13.9 Mb, supplementary data table 2), yielding a parametric LOD score of 2.3. This region contained a total of 111 genes. The selection of genes with a putative mitochondrial function was based on the MitoP2 database (http://www.mitop.de:8080/mitop2/) and the list of Calvo et al,40 which is based on a variety of criteria, including mitochondrial import sequence and co-expression with known mitochondrial genes. A total of eight candidate genes—IDH3B (isocitrate dehydrogenase 3 [NAD+] β-subunit), MRPS26 (mitochondrial ribosomal protein S26), PANK2 (pantothenate kinase 2), CRLS1 (cardiolipin synthase 1), CDS2 (CDP-diacylglycerol synthase (phosphatidate cytidylyltransferase) 2), HAO1 (hydroxyacid oxidase (glycolate oxidase) 1), PAK7 (p21(CDKN1A)-activated kinase 7), and C20orf7—were selected for mutation analysis.
Mutation detection in eight candidate genes
Exons and flanking intron sequences of the eight candidate genes were analysed by conventional sequence analysis. Only two potentially pathogenic variants were found in the CRLS1 and C20orf7 gene, while in the other six genes no pathogenic mutations were identified. Patient IV11 was homozygous for a c.592C→T mutation in exon 4 of the CRLS1 gene, changing proline at position 193 into leucine. Patient IV7 was homozygous mutant as well, both parents and two sibs were heterozygous, and one sib was wild type. The mutation was neither reported before nor present in any of the single nucleotide polymorphism (SNP) databases (NCBI SNP, Genome Variation Server and http://MutationDiscovery.com). Analysis of a Moroccan and Dutch control population (resp. 92 and 294 alleles) using sequence analysis revealed an allele frequency of, respectively, 20% and 10%, which was unexpected given the absence of the variant in the SNP databases. CLS activity was reduced to 50–56% in fibroblasts of patients compared to unrelated wild-type control fibroblasts. Based on the high frequency of the (heterozygous) mutation in the controls and the residual activity of CLS of about 50%, we concluded that this mutation could not by itself explain the rare clinical features in this family.
A second homozygous mutation was identified in the C20orf7 gene. Both patients (IV7 and IV11) were homozygous for an A-to-C transversion at nucleotide 477 in exon 5 of isoform 1 (NM_024120.3), whereas both parents and two other sibs were heterozygous and one sib was wild type (figures 1 and 3a). Two isoforms of C20orf7 are currently known, consisting of, respectively, 345 amino acids (isoform 1 (NM_024120.3), containing exons 1–11), and 317 amino acids (isoform 2 (NM_001039375.1), containing exons 1–4 and 6–11). The mutation is present in isoform 1 only. The mutation changed a leucine at position 159 to phenylalanine in the highly conserved (from man to Drosophila) S-adenosylmethionine (SAM) dependent methyltransferase domain of the protein (figure 3b). Analysis with the program SOPM41 shows that the secondary structure is likely to change due to breakage of an α helix in the mutant protein (data not shown). SOPM uses sequence information and similarities to predict secondary protein structures. The mutation was not present in 110 Moroccan and 312 Dutch alleles, supporting a pathogenic role for the mutation.
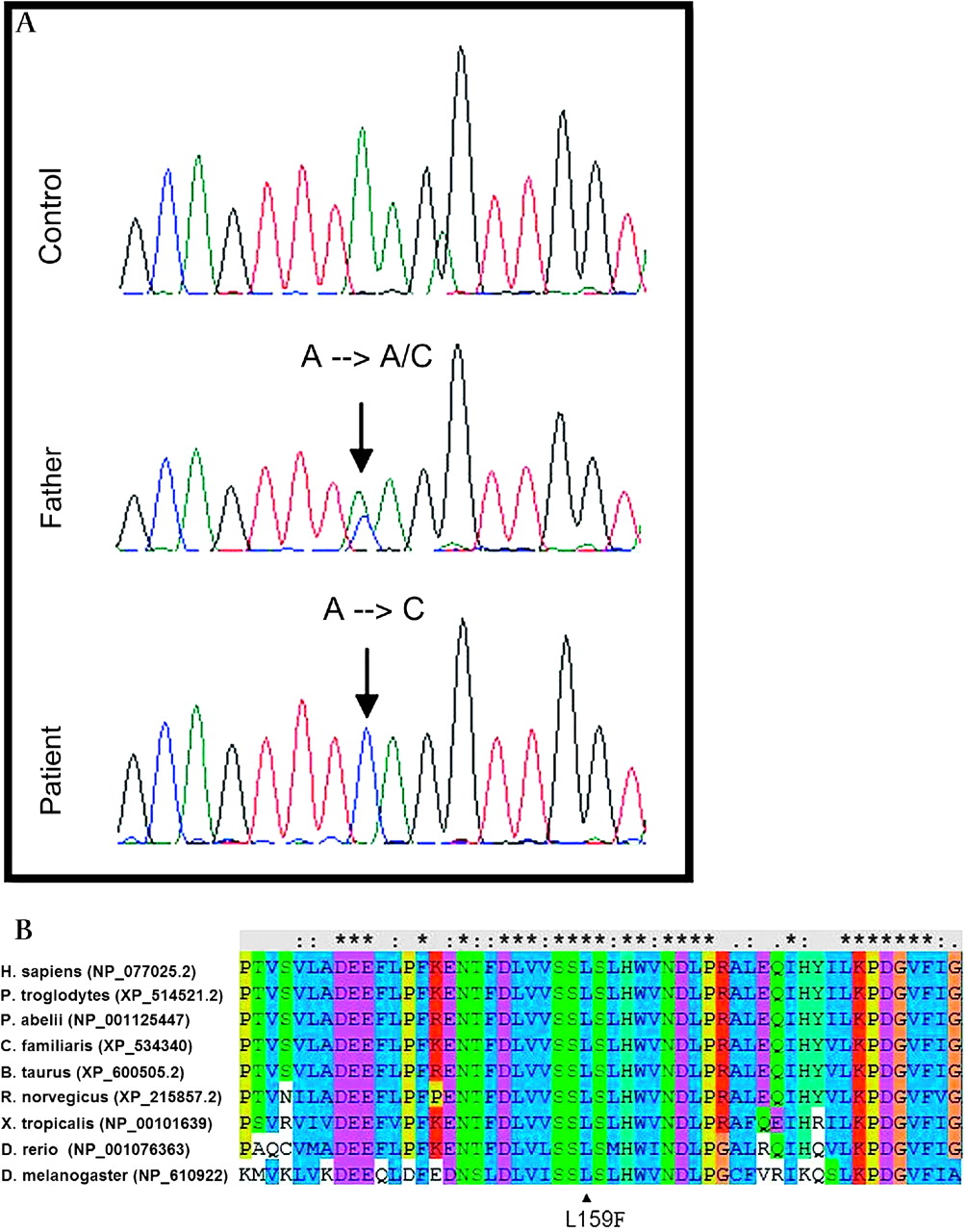
Mutation analysis in the C20orf7 gene reveals a c.477A→C mutation changing leucine at position 159 to phenylalanine. (A) Sequence analysis of exon 5 of the C20orf7 gene. (B) Conservation of leucine 159 from man to Drosophila (ClustalW).
p.L159F substitution in C20orf7 disturbs complex I assembly
Recently, Sugiana et al31 reported that C20orf7 resides within the mitochondrial matrix with a function in the assembly or stability of an early complex I assembly intermediate that contains among others ND1. Therefore we tested if the p.L159F substitution affected complex I assembly. BN-PAGE (Blue-Native polyacrylamide gel electrophoresis) was performed in PBMCs of patients IV7 and IV11 (homozygous mutant), heterozygous carriers III1, III2 and IV6 and the homozygous wild type sib IV10 with antibodies against ND1 and NDUFA9 to detect the mature 880 kDa complex I. BN-PAGE showed a decrease of mature complex I in patient samples IV7 and IV11 to 30–40% of the control values. In carriers (III1, III2 and IV6) this was 70–90% of the normal amount of complex I (figure 4A,B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Complex I assembly is altered in patient PBMC. Mitochondria were isolated from PBMC of wild-type (A/A), carriers (A/C) and a patient (C/C) donors, solubilised in 1% Triton X-100, and subjected to BN-PAGE and Western immunoblot analysis and quantified. Complex I detected by an antibody to the 39 kDa subunit (NDUFA9) or ND1 subunit is at approximately 880 kDa, and complex II detected by an antibody to the 70 kDa subunit is at approximately 130 kDa. (A) After 10–30 s exposure time. (B) Histogram of A showing the density volumes of complex I normalised by complex II as percentage of the wild-type (IV:10) donor, with error bars representing the SEM of three or four gels. *p<0.05 lower than the wild-type donor (IV:10) as compared by Student t test.
Discussion
Leigh syndrome is a progressive neurodegenerative disorder, which can be caused by mutations in the mtDNA and a variety of known and still unknown nuclear genes. Due to this genetic heterogeneity it is often difficult to establish a genetic diagnosis and characterise the pathological process involved. This study reports an early complex I assembly defect due to a missense mutation in the new complex I assembly factor C20orf7 as the pathological cause. The c20orf7 mutation affects the highly conserved S-adenosylmethionine (SAM) dependent methyltransferase domain, which may be involved in methylation of NDUFB3 as a critical step in early complex I assembly as postulated by Sugiana et al.31 The patients do not display the full picture of Leigh syndrome and clinically the phenotype has overlap with infantile bilateral striatal necrosis (IBSN, MIM 271930), but the involvement of structures in the brain stem make it more compatible to Leigh syndrome. Defects in complex I assembly/stability leading to enzymatic deficiency have been reported before in Leigh syndrome due to mutations in structural or accessory subunits.42 To our knowledge this is, next to c8orf38,31 the second defect in a genuine complex I assembly factor in Leigh syndrome, which adds defective complex I assembly as an important pathological concept to the growing list of pathogenic causes of Leigh syndrome.
When these studies were in progress, a family with a lethal neonatal form of complex I deficiency with a mutation in the C20orf7 gene was reported.31 Patients were homozygous for a missense mutation in exon 7 of the C20orf7 gene (NM_024120.3), leading to a substitution of leucine 229 to proline, which is present in both isoforms. Patients' fibroblasts almost completely lacked the mature complex I holo-enzyme. ND1 or an intermediate complex containing ND1 was not present, suggesting an early assembly defect. In concordance with their observation, a decrease of mature complex I relative to complex II down to 30–40% compared to normal is observed in our patients' PMBCs as determined by BN-PAGE.
The clinical manifestation of the p.L159F mutation in this family is less severe than the neonatal lethal p.L229P mutation, and also the complex I deficiency and assembly defect are less pronounced. Either the mutation or the affected domain of C20orf7 is less essential for proper assembly or the defect may be partially rescued in our patients by its absence in isoform 2. As no difference was observed in the relative expression of the two isoforms in different tissues, including brain, liver, heart, skeletal muscle, kidney, adipose tissue, thyroid, blood and endothelium (data not shown), it is unlikely that tissue specific expression of the two isoforms can explain the variation in clinical expression. It remains unclear whether the second isoform leads to a protein with the same function as isoform 1. Further studies are required to resolve the exact pathological or rescue process occurring.
Patients were not only homozygous for the mutation in C20orf7, but also for a common variant in the CRLS1 gene. As 1% of the Dutch and 4% of the Moroccan population is predicted to be homozygous for this mutation, this would imply a disease frequency of 1–4 in 100, if the CRLS1 variant would be the genetic cause. This is not the case. However, the reduction of CLS activity, which is unlikely to cause clinical symptoms by itself, may contribute to the disease phenotype in case the OXPHOS is already affected by another defect, like in this family. So, C20orf7 mutations are a new cause of Leigh syndrome due to an early complex I assembly defect. Disease manifestations are mutation specific and may be modified by additional mutations in other genes, like CRLS1.
Web resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
MitoP2 database, http://www.mitop.de:8080/mitop2/
NCBI SNP database, http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp
Genome Variation Server, http://gvs.gs.washington.edu/GVS/
Nucleotide Variation and Mutation Database MutationDiscovery.com, http://www.mutationdiscovery.com/md/MD.com/home_page.jsp
SOPM, http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_sopm.html
Acknowledgments
This work was supported by a EU grant to the MitoCircle project (Sixth Framework Program, contr. no. 005260). We thank Prof Dr B Oostra for supplying a Morroccon control panel.
References
Supplementary materials
Web Only Data jmg.2009.067553
Files in this Data Supplement:
Web Only Data jmg.2009.067553
Files in this Data Supplement:
Footnotes
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.