Article Text

Abstract
Background Colorectal, endometrial and upper urinary tract tumours are characteristic for Lynch syndrome (hereditary non-polyposis colon carcinoma, HNPCC). The aim of the present study was to establish whether carriers of mutations in mismatch repair genes MLH1, MSH2 or MSH6 are at increased risk of urinary bladder cancer.
Methods Carriers and first degree relatives of 95 families with a germline mutation in the MLH1 (n=26), MSH2 (n=43), or MSH6 (n=26) gene were systematically questioned about the occurrence of carcinoma. The cumulative risk of cancer occurring before the age of 70 years (CR70) was compared to the CR70 of the general Dutch population. Microsatellite instability (MSI) testing and/or immunohistochemistry (IHC) for mismatch repair proteins was performed on bladder tumour tissue.
Results Bladder cancer was diagnosed in 21 patients (90% men) from 19 Lynch syndrome families (2 MLH1, 15 MSH2, and 4 MSH6). CR70 for bladder cancer was 7.5% (95% CI 3.1% to 11.9%) for men and 1.0% (95% CI 0% to 2.4%) for women, resulting in relative risks for mutation carriers and first degree relatives of 4.2 (95% CI 2.2 to 7.2) for men and 2.2 (95% CI 0.3 to 8.0) for women. Men carrying an MSH2 mutation and their first degree relatives were at highest risks: CR70 for bladder and upper urinary tract cancer being 12.3% (95% CI 4.3% to 20.3%) and 5.9% (95% CI 0.7% to 11.1%). Bladder cancer tissue was MSI positive in 6/7 tumours and loss of IHC staining was found in 14/17 tumours, indicating Lynch syndrome aetiology.
Conclusion Patients with Lynch syndrome carrying an MSH2 mutation are at increased risk of urinary tract cancer including bladder cancer. In these cases surveillance should be considered.
- Lynch syndrome
- HNPCC
- urothelial cancer
- bladder cancer
- MSI
- gastroenterology
- clinical genetics
- genetic screening/counselling
- cancer: urological
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Lynch syndrome
- HNPCC
- urothelial cancer
- bladder cancer
- MSI
- gastroenterology
- clinical genetics
- genetic screening/counselling
- cancer: urological
Introduction
Lynch syndrome, previously called hereditary non-polyposis colon carcinoma (HNPCC), is caused by a germline mutation in one of the mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2. It results in a large increase in spontaneous mutations and thus has a direct oncogenic effect. Besides the high risk of developing colorectal carcinomas of 10–80%, Lynch syndrome family members are at increased risk of developing several extra-colonic cancers and tumours at a relatively young age: endometrial cancer, carcinomas of the ovary, small bowel and biliary tract cancer, sebaceous gland tumours and urothelial carcinomas (UC) of the upper urinary tract.1–10 The lifetime risk of upper urinary tract cancer in Lynch syndrome varies in different studies from 0.4–20%.1 2 6 9 11–18 Microsatellite instability (MSI) is present in these urothelial carcinomas of the upper urinary tract.19 20
A study based on the Swedish family cancer database, quantifying the occurrence of UC in families with at least four generations, showed that families fulfilling the Bethesda criteria21 have an increased risk of cancers in the ureter, but not in the urinary bladder.18 It was concluded that in families at risk for Lynch syndrome, UC of the bladder occurs at a frequency comparable to that in the general population. The study did not report results which were stratified by type of mutation. On the contrary, in a study by Geary et al, bladder cancer was more common in MSH2 mutation families than expected in the general population, the relative risk (RR) being 3.6 (p=0.001).16 Cumulative risks were not presented. In their study every case of bladder cancer was accompanied by cancer of the ureter in the family. For that reason, the authors concluded that the risk of bladder cancer was increased only in families with ureter cancer.
In addition to morphologic resemblance, sporadic urothelial cell cancers of the renal pelvis, ureter and bladder share the most important risk factors and molecular genetic aberrations; it is therefore remarkable that the risk of bladder cancer does not appear to have increased in Lynch syndrome families, while the risk of upper urinary tract cancer has. The aim of the present study was to establish whether patients with Lynch syndrome are at increased risk of cancer of the urinary bladder.
Patients and methods
Patient population
From 95 Lynch syndrome families, we selected all carriers of a germline mutation in the MSH2, MLH1 or MSH6 gene, who had been seen and registered at the Department of Human Genetics of the Radboud University Nijmegen Medical Centre between 1996 and November 2008, and all their first degree relatives—a total of 627 men and 617 women. Information on mutation carriers and their first degree relatives was systematically collected by postal questionnaires and telephone calls with contact persons when data were outdated for more than 1 year. In each family, one to six such persons were contacted in order to obtain up-to-date information on the occurrence of carcinoma in mutation carriers and their first degree relatives. First degree relatives with MMR gene mutation negative tests were excluded. All diagnoses of UC carcinoma were verified and confirmed by review of pathology reports or medical reports.
Statistical analysis
We calculated follow-up time for each family member with the date of birth as starting point until the dates of last contact, death, or diagnosis of cancer, whichever came first. Kaplan–Meier (KM) survival analyses were used to calculate the cumulative risk (plus SE) of cancer until specific ages. We chose the age of 70 as censuring age because the number of patients being at risk after that age is small. SPSS version 16.0 was used for KM analyses. For reference, we used life table cancer risk estimates as reported by the Dutch Cancer Registry (http://www.ikcnet.nl), based on cancer incidence and population demographics in 2003.22 The life table risks of urinary tract cancer are almost identical to those reported by SEER (Surveillance Epidemiology and End Results) for whites in the USA (http://seer.cancer.gov). Relative risks of cancer among specific groups compared to the Dutch reference risks were defined as the ratio between observed and expected numbers of tumours before the age of 70 years. Exact, 95% confidence interval (CIs) of the RRs were calculated using the publicly available OpenEpi software (http://www.openepi.com).
Histology and molecular analysis
After written informed consent, histology reports and tissue blocks of UC of the bladder, ureter or renal pelvis were collected. Clinicopathological features of the urothelial cell cancers were reviewed by two experienced pathologists using haematoxylin and eosin (H&E) slides. The following parameters were scored: (1) tumour differentiation according to the World Health Organization 2004 grading system; (2) T stage (TNM 2002); (3) growth pattern: papillary or solid; (4) presence of tumour infiltrating lymphocytes (TILs): absent, moderate or dense.
For MSI analysis, areas of the formalin fixed and paraffin embedded tissues containing either tumour cells (at least 30%) or normal cells were marked and material from these areas was isolated separately using a lysis buffer and a protein precipitation solution (Purogene, Gentra systems, Minneapolis, Minnesota, USA). PCR of the markers D2S123, D5S346, D17S250, BAT25, BAT26 and BAT40 was performed using standard conditions in the presence of a fluorescently labelled primer. GeneScan analysis was performed on an ABI373A, ABI3100 or ABI3700 apparatus (PE Biosystems, Foster City, California, USA). A tumour was considered MSI positive when at least two of these standard set of markers, or 30% of an extended set, showed instability.23
Immunohistochemistry (IHC) was performed on formalin fixed, paraffin embedded tissues. Slides were stained with antibodies against MLH1 (Pharmingen code: 51-1327gr), PMS2 (Pharmingen code: 556415), MSH2 (Oncogene Research Products code: NA26) and MSH6 (Transduction Laboratories code: G70220). Staining patterns of MMR proteins were evaluated using normal epithelial, stromal and inflammatory cells as internal controls. Stained slides were scored as: (1) positive—that is, showing nuclear staining in at least some tumour cells; (2) negative—that is, no staining of the tumour with a positive internal control; or (3) not assessable—that is, insufficient technical quality to provide an unambiguous result despite repeated assays.24 25 As a verification five anonymous sporadic cancers of the bladder were taken at random from the files of the department of pathology.
Results
Members of 95 Lynch syndrome families diagnosed by a germline mutation in MLH1 (n=26), MSH2 (n=43) or MSH6 (n=26) were questioned by contact persons for occurrence of cancer such as UC of the bladder, ureter or renal pelvis. The total cohort of 1244 persons consisted of 406 proven mutation carriers (individuals with a pathogenic mutation on germline mutation analysis) and 838 first degree relatives. The latter group consisted of 111 obligate mutation carriers (concluded because of their position in the pedigree in relation to relatives tested positive for a mutation) and 727 persons at 50% risk to be mutation carriers. In 22 families, at least one (n=27) confirmed mutation carrier or first degree relative of a mutation carrier was diagnosed with UC of the urinary bladder, ureter or renal pelvis. In two additional MSH2 families, the patient with UC of the bladder was not a first but a second degree relative of the mutation carrier diagnosed. Second degree relatives were not included in the KM risk analysis but the two second degree relatives with UC were included in the histopathology study. UC of the bladder was diagnosed in 21 patients from 19 Lynch syndrome families, the majority (71%) coming from MSH2 families (15 MSH2, two MLH1, and four MSH6); 19 out of 21 (90%) were men. Two patients with bladder cancer were at 50% risk of being an MSH2 mutation carrier, but could not be tested. Bladder cancer was diagnosed at an average age of 60±12 years (range 41–84 years), which is approximately 10 years younger than the average age of sporadic bladder cancer in the Netherlands.
The cumulative risk of bladder cancer until the age of 70 (CR70) in all MMR mutation carriers and their first degree relatives was 7.5% (95% CI 3.1% to 11.9%) for men and 1.0% (95% CI 0% to 2.4%) for women (table 1). The corresponding CR70 for the Dutch population is 1.8% for men and 0.5% for women. The RR for carriers of any MMR mutation, as compared to the general Dutch population, was 4.2 (95% CI 2.2 to 7.2, p<0.001) for men and 2.2 (95% CI 0.3 to 8.0, p=0.5) for women (table 1). MSH2 mutation carriers and first degree relatives showed the highest risk of bladder cancer: CR70 is 12.3% (95% CI 4.3% to 20.3%) in men and 2.6% (95% CI 0% to 3.9%) for women. The RR of bladder cancer in carriers of an MSH2 mutation and their first degree relatives, as compared to the general Dutch population, was 7.0 (95% CI 3.4 to 13.0, p<0.001) for men and 5.8 (95% CI 0.5 to 16.1, p=0.15) for women. The overall CR70 risk for urinary tract cancer, including the bladder, in MSH2 mutation carriers and first degree relatives was 18.2% (95% CI 5.0% to 31.4%) in men and 8.4% (95% CI 0% to 15.4%) in women (table 1).
Life table cumulative risks of cancers before the age of 70 (%)
Two or more primary urinary tract cancers, of which one was in the bladder, were diagnosed in nine of all 21 patients with bladder cancer; in six patients these two cancers were diagnosed synchronously or within 1 year. The remaining three patients, with a metachronous combination of bladder and upper urinary tract cancer, had their bladder cancer diagnosed earlier in life than the upper urinary tract cancer. Exclusive upper urinary tract cancer was diagnosed in eight patients from seven Lynch syndrome families, with 63% caused by MSH2 (five MSH2, two MLH1, and one MSH6). The CR70 of colorectal cancer for confirmed MMR mutation carriers was 63.2% (95% CI 56.6% to 69.8%) and the CR70 of endometrial cancer was 35.4% (95% CI 26.8% to 44.0%) (table 1).
Bladder cancers from germline MSH2 mutation carriers were tested for mismatch repair deficiency: MSI was present in bladder tumour DNA from six out of seven cases, and IHC staining of MSH2 protein was absent in nine out of 11 cases, indicating mismatch repair deficiency (figure 2, table 2). Additionally, IHC staining of MLH1 or MSH6 proteins was absent in five out of six bladder carcinomas from MLH1 or MSH6 germline mutation carriers, respectively (figure 1, table 2). Three bladder tumours were identified having normal IHC staining and one tumour was MSS. As a control, IHC of five sporadic bladder carcinomas showed normal staining of all four MMR system proteins. No typical histological characteristics were observed in the bladder carcinomas when compared with sporadic bladder carcinomas (table 2).
Clinico-histopathological features of all patients with urothelial carcinomas of the urinary bladder

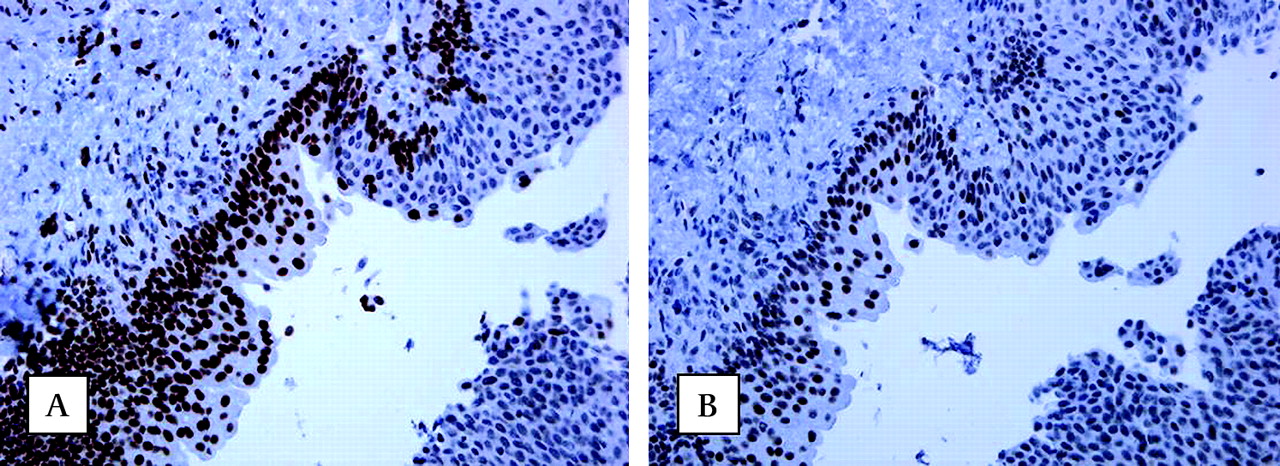
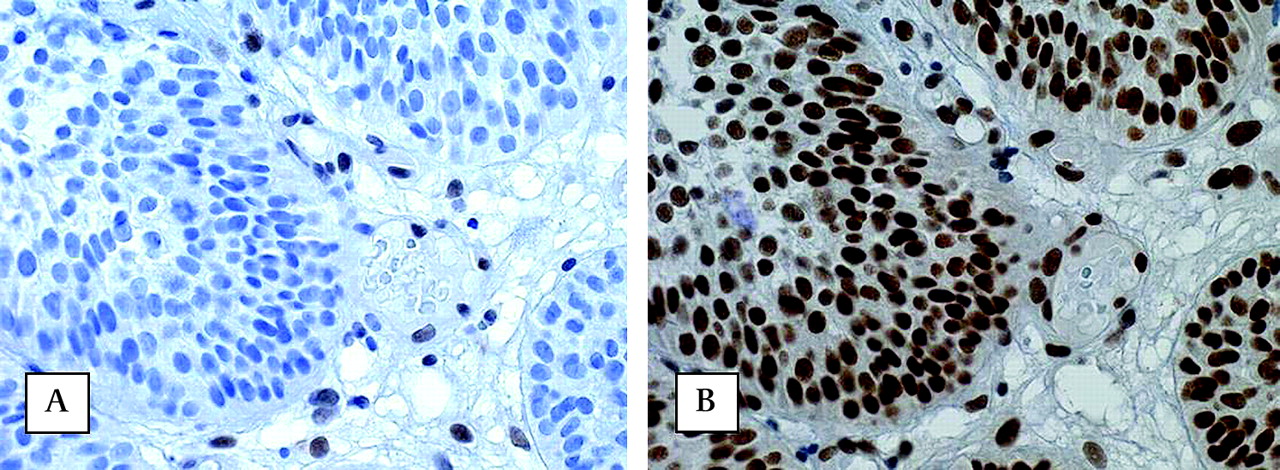
Absence of nuclear immunohistochemical staining of MLH1 protein (A) and presence of MSH2 protein (B) in urothelial carcinomas (UC) of the urinary bladder (original magnifications, 250×) of a patient carrying a germline MLH1 mutation. Note in figure 1A the presence of nuclear staining in normal mucosa and absence in carcinoma.

Absence of nuclear immunohistochemical staining of MSH2 protein (A) and presence of MLH1 protein (B) in urothelial cell carcinoma of the urinary bladder (original magnifications, 400×) of a patient carrying a germline MSH2 mutation. Observe the nuclear staining in stromal cells as an internal control.
Discussion
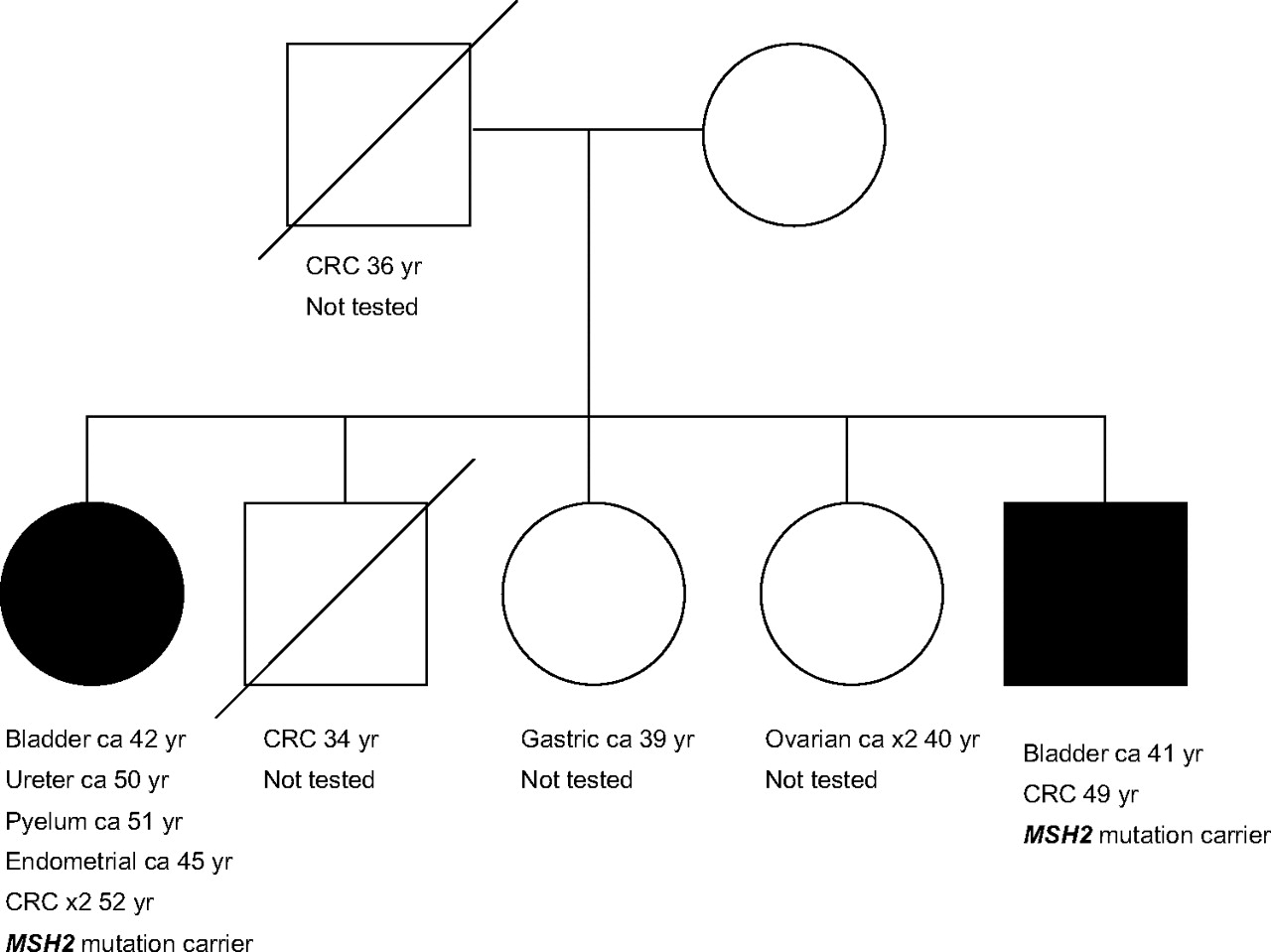
This study indicates an increased risk of urothelial cancer of both the urinary bladder and the upper urinary tract (ureter and renal pelvis) in patients with Lynch syndrome carrying a germline MSH2 mutation. Furthermore, this study indicates that cancer of the urinary bladder, ureter and renal pelvis is also, though rarely, associated with MSH6 or MLH1 mutations. A causal relation between MSH2 deficiency and bladder cancer is likely: in the first place, because bladder cancers in these families often show MSI and/or absence of IHC staining of the MSH2 protein, just like upper urothelial tract cancer and colorectal cancer; in the second place, because of the presence of two MSH2 mutation families, each of which contained two first degree family members with bladder cancer, and with three of the bladder cancers diagnosed under the age of 50 (as illustrated in figure 3). It is interesting that especially MSH2 mutation carriers are at increased risk of urothelial cancer, as observed in this cohort and in previous studies.2 12 16 The diversity in the function of the MSH2, MLH1 and MSH6 protein might be responsible for the variation in cancer risk. Environmental factors (eg, smoking status) may also affect the urologic tract cancer occurrence rates, but we do not have the necessary information to determine if this might contribute to the differences.
{kind=link}
{kind=link}
{kind=link}
Pedigree of a Lynch syndrome family with two MSH2 mutation carriers with urinary bladder cancer. CRC, colorectal cancer.
In eight out of 21 patients with bladder cancer, this was their first cancer diagnosis, whereas at this stage five of them developed another Lynch syndrome associated cancer at an older age. Therefore, early diagnosis of Lynch syndrome may prevent development of a second primary cancer, especially colorectal cancer (CRC) by regular colonoscopy with polypectomy.26 The diagnosis of Lynch syndrome in healthy relatives may lead to prevention or early detection of cancer, which improves the prognosis.26 Thus bladder cancer, just like upper urothelial tract cancer, can be added to the Bethesda criteria21 and the Amsterdam II criteria,27 and lead to the clinical suspicion of Lynch syndrome. The occurrence of bladder cancer can be used in family history taking and molecular diagnostics to identify the families that are at risk of Lynch syndrome.
Cumulative risks of colorectal and endometrial cancer are within the ranges published by other studies.2–5 8 13 28 29 Bladder cancer risk observed in our cohort was significantly higher than that observed in previous studies.6 9 12 16 18 These studies differed from our study by: (1) type of risk that was calculated; (2) by type of population; and (3) whether or not the type of mutation was distinguished. The data from these studies are given in table 3. Additionally the discrepancy in bladder cancer risk between our study and other studies may result from our systematic data collection approach (obtaining up-to-date information). This led to the discovery of nine new cases of bladder carcinoma, previously unrevealed with the standard procedure of a clinical geneticist taking the family history.
Overview of risk of bladder cancer in previous studies
Considering the high risk of urothelial tract cancer in MSH2 mutation carriers, a surveillance programme needs to be developed. At present various recommendations have been published. The present European guidelines for families with an MMR mutation include ultrasound and urinalysis every 1–2 years with patients from the age of 30 to 35 only in cases where upper urinary tract cancer runs in the family (two or more cases of UC).11 The American guidelines include urinalysis with cytology every 1–2 years with patients from the age of 25 to 35 for all family members with Lynch syndrome.30 Watson et al proposed (unspecified) surveillance of patients starting with the age of 50, especially for families that carry mutations in MSH2.12 Although cytology is the superior marker in terms of specificity,31 Myrhoy et al showed that the sensitivity of urine cytology in diagnosing asymptomatic upper urinary tract cancer in Lynch syndrome is approximately 30%. Therefore, cytology only is not a proper method of surveillance.32 The most important biomarker of urothelial cancer is macro- or microscopically haematuria, which occurs in 85% of patients with bladder cancer.33 Consequently, Koornstra et al recommend annual surveillance for haematuria, by urinary dipstick, of all patients with Lynch syndrome, beginning at the age of 45 to 50.34 The recent use of sensitive transducers has improved imaging of the upper urinary tract and bladder by ultrasonography. It was shown to be as accurate in the detection of renal masses and bladder filling defects as intravenous urography and computed tomography scanning.35
Further studies are needed to develop an optimum early detection strategy concerning the appropriate interval, methods to be used, and patient groups to be included. Until then, we propose a combination of ultrasonography of the bladder and upper urinary tract, urinary cytology and urine sediment (erythrocytes) every 1–2 years. We recommend a surveillance programme for UC of the bladder and upper urinary tract for all MSH2 mutation carriers starting at the age of 40, which is based on the youngest Lynch syndrome patient with bladder cancer reported in the literature (age 40) and observed in our study (age 41).36 The top age limit of surveillance for UC can be similar to that of surveillance for colorectal cancer. Surveillance should not be limited to families with a history of UC, because in our study clustering of urinary tract cancer was only observed in five families, while 19 patients had a negative family history of urinary tract cancer.
In conclusion, our data suggest that in addition to upper urinary tract cancer, urothelial cancer of the urinary bladder is part of the Lynch syndrome tumour spectrum. Carriers of an MSH2 mutation are particularly at increased risk of urinary tract cancer including cancer of the bladder. In these cases we consider surveillance necessary.
Recommendations for urothelial carcinomas surveillance in Lynch syndrome
Surveillance with a combination of ultrasound of the bladder and upper urinary tract, urinary cytology and sediment.
In every MSH2 mutation carrier
From age 40 and up
Performed every 1–2 years
Acknowledgments
We would like to thank Irene Reimerink and Marsha Voorendt (genetic counselors), Dr Ernie Bongers (clinical geneticist), Riki Willems, Monique Link, Monique Goossens, Sandra Hendriks-Cornelissen and Evelien Hoenselaar (technical assistance).
References
Footnotes
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.