Article Text

Abstract
Background The introduction of molecular karyotyping technologies facilitated the identification of specific genetic disorders associated with imbalances of certain genomic regions. A detailed phenotypic delineation of interstitial 16p13.3 duplications is hampered by the scarcity of such patients.
Objectives To delineate the phenotypic spectrum associated with interstitial 16p13.3 duplications, and perform a genotype-phenotype analysis.
Results The present report describes the genotypic and phenotypic delineation of nine submicroscopic interstitial 16p13.3 duplications. The critically duplicated region encompasses a single gene, CREBBP, which is mutated or deleted in Rubinstein–Taybi syndrome. In 10 out of the 12 hitherto described probands, the duplication arose de novo.
Conclusions Interstitial 16p13.3 duplications have a recognizable phenotype, characterized by normal to moderately retarded mental development, normal growth, mild arthrogryposis, frequently small and proximally implanted thumbs and characteristic facial features. Occasionally, developmental defects of the heart, genitalia, palate or the eyes are observed. The frequent de novo occurrence of 16p13.3 duplications demonstrates the reduced reproductive fitness associated with this genotype. Inheritance of the duplication from a clinically normal parent in two cases indicates that the associated phenotype is incompletely penetrant.
- Microduplication
- CREBBP
- Rubinstein–Taybi syndrome
- genotype–phenotype correlation
- arthrogryposis
- genetics
- cytogenetics
Statistics from Altmetric.com
- Microduplication
- CREBBP
- Rubinstein–Taybi syndrome
- genotype–phenotype correlation
- arthrogryposis
- genetics
- cytogenetics
Introduction
Chromosomal imbalances are a cause of genetic disorders and often cause a syndromic phenotype. The identification of such imbalances on a genome-wide level was until recently only feasible by investigating metaphase chromosomes under a microscope. The latest developments in microarray technology, however, enable assessment of copy number of thousands to millions of loci across the human genome at a resolution far surpassing that of conventional karyotyping. The introduction of these technologies into the diagnostic work-up of patients with congenital disorders represents a revolution in this field, the importance of which cannot be overstated. It led to a vast improvement in the aetiological diagnosis of patients with previously idiopathic congenital abnormalities, mental retardation (MR) and/or psychiatric problems.1 It moreover permitted the identification and delineation of novel microdeletion and microduplication syndromes, thus allowing a more detailed assessment of the phenotypic consequences associated with specific chromosomal imbalances and more accurate and targeted care and counselling of patients and their parents.2 The present study describes the identification and delineation of a novel microduplication syndrome, microduplication 16p13.3, which is complementary to microdeletions of 16p13.3 that cause Rubinstein–Taybi syndrome (RTS).3
Patients, materials and methods
Patients described in this study were referred for idiopathic MR and/or congenital anomalies. They were followed in the collaborating centres, and clinical data and informed consent were obtained from all patients or their legal representatives. A genome-wide copy number profile of the patient DNA was obtained by subjecting it to microarray analysis using the Affymetrix 500K GeneChip platform (patient 2), the genome-wide 105K V7 OLIGO array (a custom designed array manufactured by Agilent Technologies Inc (Santa Clara, California, USA), containing oligonucleotides as probes chosen genome-wide with an average interval of 30 kb plus an enrichment of probes in most known regions associated with syndromes, pericentromeric and subtelomeric regions) (patients 1, 3, 6 and 8), the Agilent 244K platform (patients 7, 9 and 11) or the Agilent 44K platform (patient 12), all according to manufacturers' recommendations, or using a tiling resolution bacterial artificial chromosome (BAC) array of chromosome 16 (patient 4) as described.4 The duplication in patient 9 was first detected by a microdeletion multiplex ligation dependent probe amplification (MLPA) kit (MRC Holland, Salsa MLPA P245) (Amsterdam, the Netherlands). All genome coordinates mentioned in this study are according to human genome build 18 (NCBI 36.1). The inheritance of each of these duplications was investigated by analysing parental samples using microarrays (patient 2) or targeted approaches such as fluorescent in situ hybridisation (FISH) (patients 1, 3, 4, 6, and 8), MLPA (patients 9 and 11) or quantitative polymerase chain reaction (PCR) (patients 7 and 12). Positions of segmental duplications were downloaded from the segmental duplication database website (http://humanparalogy.gs.washington.edu/build36/). The duplications in patients 2, 5 and 10 were previously reported.5–7
Results
Nine patients with an interstitial duplication of 16p13.3 are described for the first time in the present study. Of the three previously reported patients (patients 2, 5 and 10),5–7 we have added more accurate genotypic and phenotypic data for patient 2. The results from micro-array analyses for patients 1–4, 6–9 and 11–12 are depicted in figure 1 together with the reported results from investigations in patients 5 and 10. The molecular data are summarised in table 1. The duplication occurred de novo in 10 cases and was inherited from a clinically normal parent in two cases (patient 3 (maternal) and patient 8 (paternal)).
Genotypic and phenotypic characteristics of patients with an interstitial 16p13.3 duplication

Upper panel: the extent of the duplications identified in the present study. Light grey bars indicate normal copy number, black bars indicate duplicated regions, dark grey bars indicate the breakpoint containing regions. Patients are ordered according to the telomeric breakpoint of the duplication. Lower panel: a view of the University of California Santa Cruz (UCSC) genome browser (NCBI build 36.1, March 2006), illustrating the genes mapped to the implicated region (first track), the regions that are copy number variable in the normal population (Database of Genomic Variants, second track), and the regions that have paralogues in the human genome (Segmental duplication – Seg Dup; third track). The transparent grey box demarcates the smallest region of overlap. All breakpoints appear unique and do not map specifically in low copy repeats.
Comparing the extent of all duplicated regions enabled the delineation of a critically duplicated region, which contains a single gene: CREBBP (figure 1). We also reanalysed the molecular karyotype of the patient reported to have an interstitial 16p13.3 duplication by de Ravel,8 using a higher resolution platform (Agilent 244K). This revealed that the duplication described in this patient was not interstitial but terminal, extending from 0 kb to 8633–8648 kb, and is 2–10 times larger than the interstitial duplications in patients 1–12. This patient was, therefore, not included in the present analysis.
There was no family history of birth defects except for patient 2, whose father had one brother with a congenital heart defect who died at 8 months of age and one sister with a history of cardiac arrhythmias, and patient 3, where both parents had heart murmurs at birth that resolved spontaneously after a few years.
Phenotypically, interstitial duplications of 16p13.3 are associated with variable mental development (ranging from normal to moderately delayed), and occasional behavioural problems such as attention deficit hyperactivity disorders, aggressive behaviour, and autism spectrum disorders. Pregnancy was uncomplicated in most; the mother of patient 3 had one abnormal non-stress test and mild oligohydramnios was noted, and patient 12 had intrauterine growth retardation. There was no exposure to alcohol or other known teratogens except in patient 3, where the mother reported occasional alcohol consumption in the first weeks of pregnancy. Birth was at term in all patients. Growth was normal in all individuals except patient 12 who had precocious puberty.
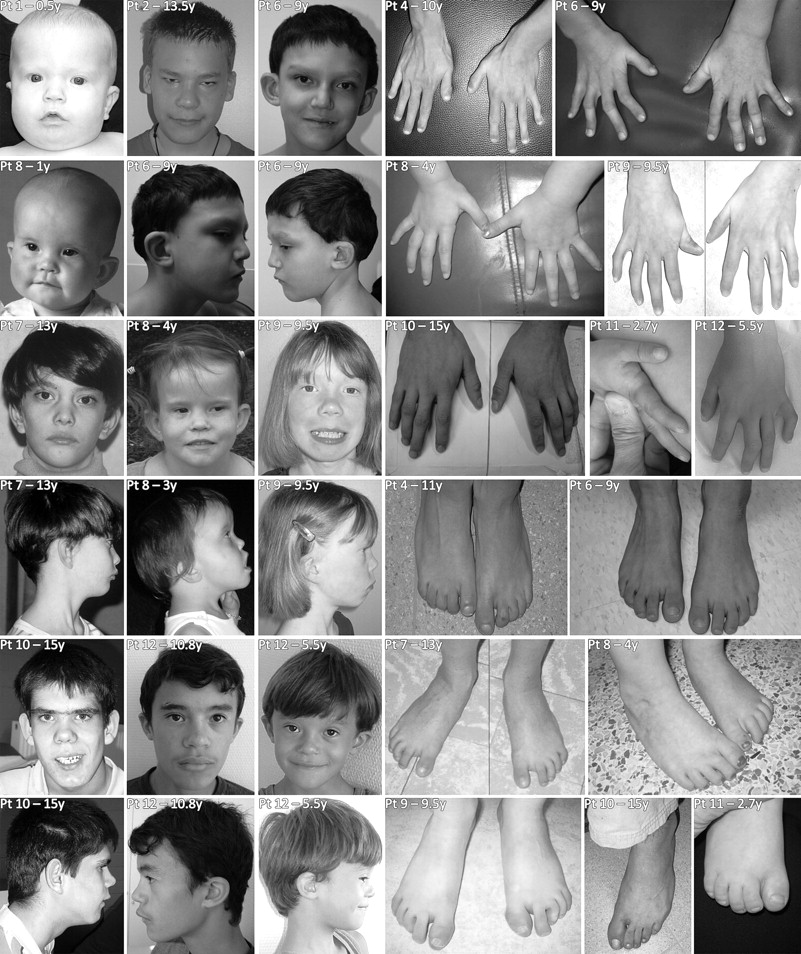
Many patients shared similar facial features. There was midfacial hypoplasia in young children and a longer face in older individuals. The nose was prominent and had a bulbous tip, and the eyes were often upslanting with narrow palpebral fissures, sometimes with ptosis. The upper lip typically was thin, and the ears low set and/or protruding (figure 2). We frequently observed mild abnormalities of the hands (the thumbs were often proximally implanted and short, fingers were long and tapering, the fifth finger was often short, there was also often camptodactyly or mild cutaneous syndactyly) and of the feet (club feet, camptodactyly, or syndactyly) (figure 2). Less frequent were other anomalies of the skeleton (congenital hip dislocation, vertebral fusion), the eyes (blepharophimosis, epicanthus inversus, strabismus, astigmatism, or ptosis), and the heart (atrial septal defect, tetralogy of Fallot). Occasional findings include inguinal hernia (twice), precocious puberty, cryptorchidism (twice), submucosal cleft palate, and mild periventricular heterotopia on magnetic resonance imaging (MRI) examination of the brain. An overview of the phenotypic data of the individual patients is given in table 1. The frequent occurrence of features such as club feet, camptodactyly of the toes and fingers, congenital hip dislocation, and incomplete extension of the elbows is indicative of mild arthrogryposis in these patients.

{kind=link}
{kind=link}
Phenotypic features of patients 1, 2, 4 and 6–12. Please note that patient 2 has had a surgical correction for ptosis.
A total of about 25 700 patients have been analysed for duplication of the CREBBP gene in our centres. In this patient population, 11 duplications of CREBBP were found, suggesting a frequency of this duplication of around 0.043% in these patients. As about 2–3% of live births fit the inclusion criteria for our patient cohort (multiple congenital anomaly/mental retardation (MCA/MR)) and about 80% of these are idiopathic before molecular karyotyping,9 we estimate the frequency of this duplication to be 1 in 97 000 to 146 000 live births.
Three different mechanisms have been proposed to generate chromosome imbalances: non-allelic homologous recombination (NAHR), non-homologous end joining, and fork stalling and template switching (FoSTeS).10 When a region is recurrently found to be deleted as well as duplicated, NAHR has been suggested as a potential underlying mechanism. However, similar to the breakpoints of deletions in this region, all breakpoints of duplications appear unique, arguing against a mechanism of non-allelic homologous recombination. To further exclude a local homology based molecular mechanism, we assessed the presence of segmental duplications (SD; length >10 kb, over 90% homology) in the breakpoint containing regions. SDs encompass 7.7% of the region between 1800–6500 kb, but none of the breakpoints falls clearly within an SD, and only two out of 22 breakpoint containing regions (9%) encompass an SD. We therefore exclude NAHR as the mechanism generating these imbalances. The lack of recurrent breakpoints for the copy number variations in these cases indicates other mechanisms, such as non-homologous end joining or FoSTeS, as possibly causative. There were no complex imbalances identified, a hallmark of the recently proposed FoSTeS mechanism, although the resolution of the analysis was probably insufficient to assess this critically in most cases. None of the parents reported exposure to known mutagens.
Discussion
The present report details genotype and phenotype of nine patients with an interstitial 16p13.3 duplication. Three patients that were previously reported were also included,5–7 with more precise genotypic and phenotypic data for one of these (patient 2).6 Careful assessment of the phenotypic features of these individuals enabled the description of a characteristic phenotype, with normal to moderately retarded mental development, mild arthrogryposis-like anomalies of the musculoskeletal system (club feet, congenital hip dislocation, or camptodactyly of the fingers and toes), mild facial dysmorphism that changes with age and occasional anomalies of the heart (atrial septal defect, tetralogy of Fallot).
It is noteworthy that all patients were selected for study because of MR and/or congenital anomalies. There might thus be—as in most genetic disorders—a bias towards the more severe end of the spectrum in the described phenotype; normal or mildly affected individuals do not present at the genetics clinic. In two instances the duplication was indeed found to be inherited from an apparently normal parent (patients 3 and 8) who had both followed normal schooling, function normally in society, and do not present the typical face. However, the frequent de novo occurrence (in 10 of 12 patients) indicates that this duplication is associated in most cases with a reduced reproductive fitness. In line with this is the finding that CREBBP duplications were never identified in genome-wide copy number profiling studies of more than 4000 individuals recruited from normal control populations.11–13 In the patient populations we studied, we identified an interstitial 16p13.3 duplication in 11 out of 25 700 individuals, suggesting that it occurs in about 0.043% of patients with MR or birth defects.
Three typical patients (9, 10 and 11) carry a very small duplication (maximal sizes 356, 480 and 639 kb), indicating that this is the critical region associated with the described phenotype. This small region encompasses only six genes. Of these genes, CREBBP is the most attractive candidate gene. The smallest region of duplication overlap (186–260 kb in size) in the 12 described individuals contains only the CREBBP gene. The distally flanking gene, ADCY9, is unaffected in patient 2. The proximally flanking gene, TRAP1, is only partially duplicated in patient 12, rendering the extra copy probably not functional. CREBBP encodes a histone acetyl transferase and thus functions as a transcriptional co-activator by decondensing chromatin and activating gene transcription. Heterozygous loss-of-function mutations and deletions of this gene have been shown to cause RTS,3 14 demonstrating that human development is sensitive to CREBBP dosage.
None of the recurrent phenotypic findings in our patients associate specifically with a region proximal or distal to CREBBP except for ptosis, which was found only in three patients with a more telomerically extending duplication (patients 2–4). In contrast to duplications extending to the subtelomere,15 interstitial duplications are not associated with microcephaly or growth retardation. Other features reported for terminal duplications of 16p seem to associate with duplication of CREBBP. These include heart defects, MR and the arthrogryposis-like features.15
Reciprocal duplications are now being clinically delineated for many of the previously described microdeletion syndromes. Examples include 7q11.23 duplications (reciprocal to Williams–Beuren syndrome deletions),16 15q11.2 duplications (reciprocal to Prader–Willi and Angelman syndrome deletions),17 17p11.2 duplications (known as Potocki–Lupski syndrome, reciprocal to Smith–Magenis syndrome deletions),18 22q11.2 duplications (reciprocal to DiGeorge syndrome deletions),19 20 Xq28 duplications (MECP2 duplications, reciprocal to Rett syndrome deletions)21 and 17p13.3 duplications (reciprocal to Miller–Dicker syndrome deletions).22 Some recurrent themes are emerging. In general, the phenotypic manifestations of reciprocal duplications are milder and much more variable than those of the deletions. Of interest, non-penetrance, which we documented in two cases with duplication 16p13.3, has also been reported for several of these duplications (eg, dup 22q11.2, dup 15q11.2, …).17 19
The defects observed in reciprocal deletion and duplication syndromes often involve the same organs or functions—for example, the heart in DiGeorge syndrome and duplication 22q11.2, speech in Williams–Beuren syndrome and duplication 7q11.23, the teeth in Smith–Magenis and Potocki–Lupski syndromes, or the nerves in Charcot–Marie–Tooth 1A and hereditary neuropathy with liability to pressure palsies.23 24 Similarly, defects of the hands, feet and heart are seen in both deletion and duplication of 16p13.3.
In some instances, an intriguing reciprocal phenotype is seen in the deletion versus duplication. For example, 7q11.2 deletions have high verbal functioning, compared to particular deficiencies in speech and language development observed in the duplication 7q11.2.16 The bulbous nasal tip observed in the present patients may perhaps be a reciprocal phenotype to the long columella seen in RTS, the short and proximally implanted thumbs may be reciprocal to the broad thumbs typical for RTS, and the arthrogryposis-like feature arguably compare to the joint hypermobility.25
The present findings indicate that normal CREBBP dosage is restricted not only by a lower threshold (as demonstrated in RTS) but also by an upper threshold. The described duplications most likely cause only a slight increase in CREBBP expression, with the normal genomic dose being only 33% lower than the dose upon duplication. This poses significant challenges to the recently proposed strategy to treat RTS patients with histone deacetylase inhibitors26: the dosage dependency of CREBBP suggests that such pharmaceutical interventions will prove beneficial in a very limited range of concentrations only. On the other hand, administering a precise dosage of histone acetyl transferase inhibitors such as curcumin27 presents a valuable line of investigation to develop a therapy for the novel genomic disorder produced by CREBBP duplication. Curcumin is a cell permeable inhibitor of multiple cellular targets including CREBBP and p300 (a paralogue mutated in an allelic form of RTS), which is currently being tested in clinical trials for neoplastic and immunological disorders.28 Although errors in development will not be curable by such strategies, the observed problems in mental function might benefit from this therapeutic intervention.
Acknowledgments
The authors would like to express their sincere gratefulness to the patients and parents who agreed to participate in this study. BT is supported by an IWT specialisatiebeurs; JB is an aspirant FWO; NP is supported by PHRC 04-07; JMF's work was supported by a grant from Genome Canada. This work was made possible by grants from GOA/2006/12 and Center of Excellence SymBioSys (Research Council K.U.Leuven EF/05/007).
References
Footnotes
Funding Other funders: BT is supported by an IWT specialisatiebeurs; JB is an aspirant FWO; LV is supported by grants from Inserm and ANR Gis-Maladies Rares; NP is supported by PHRC 04-07; JMF's work was supported by a grant from Genome Canada. This work was made possible by grants from GOA/2006/12 and Center of Excellence SymBioSys (Research Council K.U.Leuven EF/05/007).
Competing interests None.
Ethics approval This study was conducted with the approval of the Etisch Commitee K.U.Leuven, Belgium.
Provenance and peer review Not commissioned; externally peer reviewed.