Article Text

Abstract
Background A child with autism and mild microcephaly was found to have a de novo 3.3 Mb microdeletion on chromosome 1p34.2p34.3. The hypothesis is tested that this microdeletion contains one or more genes that underlie the autism phenotype in this child and in other children with autism spectrum disorders.
Methods To search for submicroscopic chromosomal rearrangements in the child, array comparative genomic hybridisation (aCGH) was performed using a 19 K whole genome human bacterial artificial chromosome (BAC) array and the Illumina 610-Quad BeadChip microarray. Ingenuity pathway analysis (IPA) was used to construct functional biological networks to identify candidate autism genes. To identify putative functional variants in candidate genes, mutation screening was performed using polymerase chain reaction (PCR) based Sanger sequencing in 512 unrelated autism patients and 462 control subjects.
Results A de novo 3.3 Mb deletion containing ∼43 genes in chromosome 1p34.2p34.3 was identified and subsequently confirmed using fluorescence in situ hybridization (FISH). Literature review and bioinformatics analyses identified Regulating Synaptic Membrane Exocytosis 3 (RIMS3) as the most promising autism candidate gene. Mutation screening of this gene in autism patients identified five inherited coding variants, including one (p.E177A) that segregated with the autism phenotype in a sibship, was predicted to be deleterious, and was absent in 1161 controls.
Conclusions This case report and mutation screening data suggest that RIMS3 is an autism causative or contributory gene. Functional studies of RIMS3 variants such as p.E177A should provide additional insight into the role of synaptic proteins in the pathophysiology of autism.
- autism
- microcephaly
- mental retardation
- copy number variants
- synapse
- molecular genetics
- neurosciences
- psychiatry
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- autism
- microcephaly
- mental retardation
- copy number variants
- synapse
- molecular genetics
- neurosciences
- psychiatry
Introduction
Autism is characterised by deficits in reciprocal social interaction, disrupted verbal and non-verbal communication, and restricted interests and repetitive behaviours, and is known to have a strong genetic component.1 2 Cytogenetic studies indicate that ∼3–4% of autism patients have chromosomal imbalances3–5 that include maternal 15q11q13 duplications (∼1–2% of patients) and deletions of 2q37, 22q11.2 and 22q13.3.4 6 Higher resolution approaches using array comparative genomic hybridisation (aCGH) and single nucleotide polymorphism microarrays have recently identified submicroscopic copy number variants (CNVs),7–10 including recurrent microdeletions and duplications of 16p11.2 that have been reported in ∼1% of autism patients.8 11 12 Importantly, the discovery of chromosomal abnormalities and CNVs has led to the identification of rare intragenic mutations through deep resequencing of autism candidate genes, including NLGN3 and NLGN4,13 NRXN1,10 14 15 SHANK316–18 and CNTNAP2.19–21 Although mutations in these genes account for only a small proportion of individuals with autism, they nonetheless provide insight into potential biological mechanisms that may underlie autism, such as synaptic dysfunction and abnormal brain connectivity.22 23
Here, we report the case of a boy with autism, microcephaly, and other dysmorphic features in whom aCGH detected a ∼3.3-Mb interstitial deletion in chromosome 1p34.2p34.3 that contains ∼43 genes, including Regulating synaptic membrane exocytosis protein 3 (RIMS3), a gene previously shown to be dysregulated in autism subjects.24 We examined the role of RIMS3 in 512 unrelated autism subjects by performing mutation analyses to look for putative functional variants.
Clinical report
The boy (LP99-105) was born at term to healthy and unrelated parents, and appeared normal except for right hydronephrosis and vesicoureteral reflux, which both resolved. His head circumference was normal at birth, but decelerated after 7 months and subsequently followed a curve at or just below −2 SD (figure 1A). His early motor development was normal, but he walked late at 16 months. He used his first few words at 15 months and slowly increased to 50–75 words by 2.5 years, but then progressively stopped using almost all speech by 3 years.

(A) The patient's head circumference (HC) was normal at birth, but decelerated after 7 months and subsequently followed a curve at −3 SD. Y axis shows HC size in both centimetres and inches. X axis indicates age. The top and bottom dashed curves represent HC at +2 SD (98%) and −2 SD (2%), respectively, in relation to the mean (50%) HC curve (solid). Serial HC measurements for the patient are represented as red dots. (B) The patient's facial appearance is normal at 1 year of age. (C) At 10 years the boy has subtle dysmorphism with a long narrow face and deep set or sunken eyes.
On examination, he had a normal facial appearance at 1 year (figure 1B), but by 10 years he had a long narrow face and deep set or sunken eyes (figure 1C). His general and neurological examinations were normal, including good motor coordination. Serial exams between 2 and 10 years all demonstrated poor social communication including poor eye contact and limited interactive play, striking anxiety, difficulty with transitions, constant chewing, short attention span, poor sleep pattern, and repetitive activities such as bouncing on an exercise ball. Brain magnetic resonance imaging (MRI) at 1 year demonstrated mildly prominent extra-axial spaces, but was otherwise normal. Serial audiograms done during his first several years of life demonstrated normal hearing.
He was diagnosed with autistic disorder at age 4 years at a university based assessment clinic for children with developmental disorders. On the Autism Diagnostic Observation Schedule–Generic (ADOS-G),25 he exceeded the autism cut-off score on the communication, social, and communication plus social domains, thus meeting the criterion for an Autism Diagnostic Observation Schedule–Generic classification of autism (domain scores: communication 6; social 12; stereotyped behaviours and restricted interests 4). The diagnosis was further supported by meeting the cut-off scores of autism on the Autism Diagnostic Interview–Revised (ADI-R)26 with his mother as the informant. More widespread developmental abnormalities were documented at 4 years based on an overall standard score of 28 on the Scales of Independent Behavior–Revised,27 a parent report measure of adaptive behaviour. This falls in the “limited to very limited” range, equivalent to behaviours at 18 months in typically developing children.
During the subsequent 6 years, he has not regained any language skills, except for using several signs inconsistently. He developed a striking fear response when placed in a car at age 9 years that has persisted for more than 3 years. Chromosome analysis and polymerase chain reaction (PCR) based fragile X testing were normal, and no other genetic tests were done. This boy is not part of the Autism Genetics Resource Exchange (AGRE, Los Angeles, California, USA) consortium (http://www.agre.org/).
Subjects and methods
Autism and control subjects
We obtained phenotype data and DNA from the 1p34 deletion proband under a protocol approved by the Institutional Review Board at the University of Chicago. The remaining autism probands selected for this study were a subset of the AGRE subjects that were collected with informed consent and institutional review board approval.28 The AGRE cohort consisted of 512 unrelated autism subjects (151 females and 355 males) from 84 simplex and 428 multiplex families. Subjects who missed criteria for autism on the ADI-R were classified as having ‘Not Quite Autism’ (NQA) if their scores on each ADI-R behavioural domain were within one point of the criteria, or if they met criteria for each domain except for age of onset <3 years of age. Fragile X testing was performed in all families and a subset of families had karyotyping and CNV analyses performed. Genomic DNA from control subjects (225 females and 237 males) was obtained from the National Institute of Mental Health Human Genetics Initiative control sample set. These subjects were screened for Axis I mental health disorders and none were reported as having autism. A summary of the ethnic origins of the autism patients and control subjects is provided in supplementary table 1.
Array comparative genomic hybridisation
We performed aCGH on the patient using a 19-K whole genome tiling path bacterial artificial chromosome (BAC) microarray as previously described.7 We refined the size of the 1p34.2p34.3 microdeletion using the Illumina 610-Quad BeadChip microarray (Illumina Inc, San Diego, California, USA, http://www.illumina.com/).
Fluorescence in situ hybridisation (FISH) and microsatellite analyses
FISH and microsatellite analyses were performed using standard techniques (supplementary materials).
DNA amplification and sequencing
PCR primers were designed using MIT Primer3 with M13 forward and reverse tails added to each primer to facilitate high throughput DNA sequencing (supplementary table 2). DNA amplification and purification were performed using standard conditions and protocols (supplementary materials).
Pathway analyses
A RIMS3 network was predicted through the use of Ingenuity Pathways Analysis software (Ingenuity Systems Inc, Redwood City, California, USA; http://www.ingenuity.com/). To generate this network, a dataset of known autism genes (ie, focus genes) was uploaded into the application. The Ingenuity Pathways Knowledge Base was used to identify interactions between the focus genes and other gene objects. The network was then algorithmically generated based on their connectivity. The network was comprised of ‘nodes’—that is, the gene or gene product, connected by ‘edges’ that are biological relationships supported by at least one publication. The network was then trimmed to show a refined network with minimal connections between the autism focus genes.
Protein predictions
We used several applications to predict the effects of missense substitutions on protein function including SIFT (Sorting Intolerant From Tolerant, http://sift.jcvi.org/),29 PolyPhen (Polymorphism Phenotyping, http://coot.embl.de/PolyPhen/),30 and SNAP (http://cubic.bioc.columbia.edu/services/snap/).31
Results
Discovery of a 1p34.2p34.3 microdeletion in a child with autism and microcephaly
We performed aCGH using a 19-K whole genome tiling path BAC microarray in patient LP99-105, and detected a ∼3.3 Mb deletion in chromosome 1p34.2p34.3 that extended from RP11-769L8 to RP11-483I17 and included ∼47 genes (figure 2A). The microdeletion was not detected in 372 control subjects analysed on the same BAC array platform.7 FISH studies confirmed the deletion in the proband (figure 2B), and microsatellite analysis demonstrated that the deletion was de novo (data not shown). We used the Illumina 610-Quad BeadChip microarray to refine the breakpoints to approximately chr1:39,794,296 and chr1:43,058,974 (UCSC Genome Browser, http://genome.ucsc.edu; Build 36.1; accessed November 2008), reducing the size of the microdeletion by ∼56 kb and identifying ∼43 genes within the deleted region.
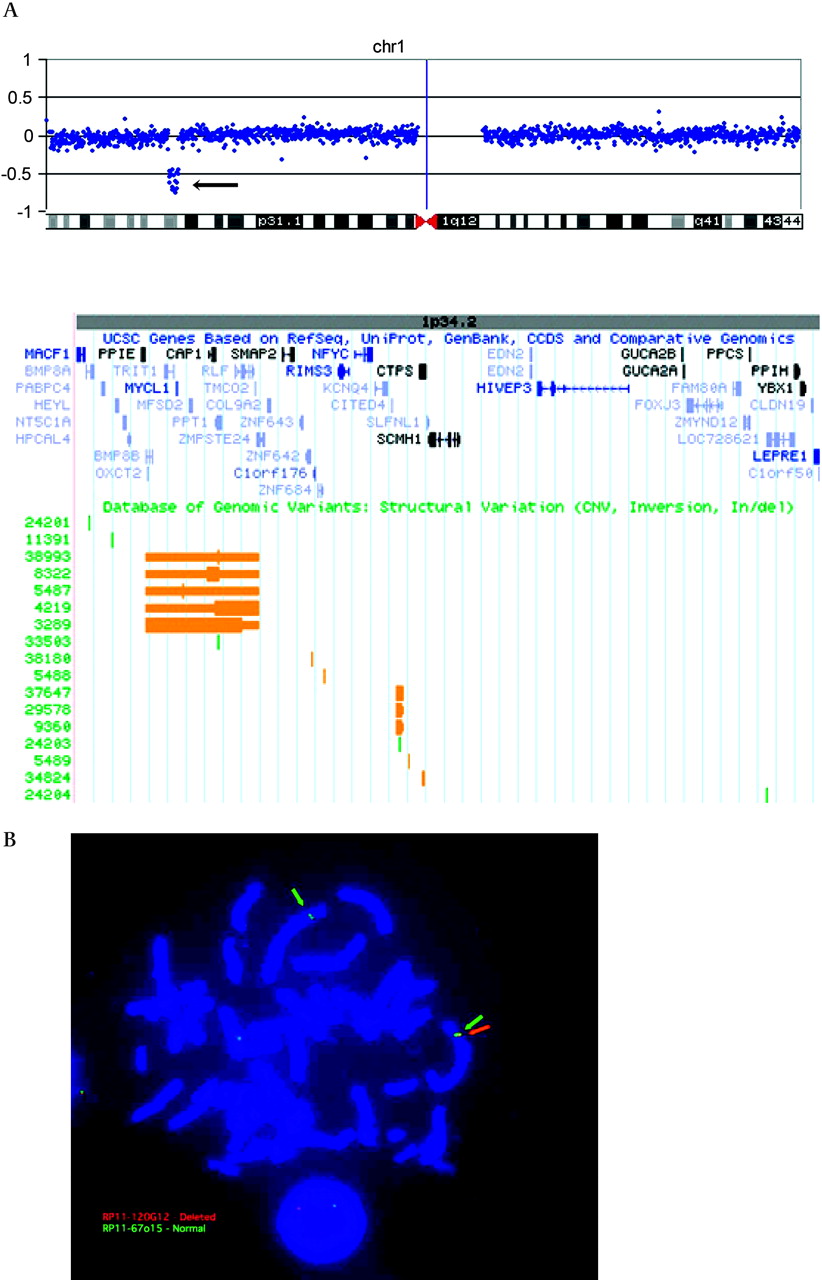
(A) Array comparative genome hybridisation (aCGH) using a bacterial artificial chromosome (BAC) microarray demonstrates a deletion in chromosome 1p34.2. The aCGH plot shows the log2 ratio of the patient versus reference DNA on the vertical axis. Each individual BAC is represented as a single blue dot and the horizontal axis shows the position of each BAC along chromosome 1. The deletion of 1p34.2 is indicated by an arrow pointing to a vertical line of dots. Interrogation of the UCSC genome browser for the microdeletion region in this region identifies ∼47 RefSeq genes (shown below aCGH plot). Known copy number variants (CNVs) (orange blocks) and Indels (green blocks) are reported in the Database of Genomic Variants track. (B) Fluorescence in situ hybridisation (FISH) analysis confirms the deletion 1p34.2 aCGH results. The distal breakpoint boundary is indicated where RP11-67o15 (green arrows) shows two normal signals while RP11-120G12 (red arrow) shows a single signal in the interphase nucleus.
We reviewed the Database of Genomic Variants (http://projects.tcag.ca/variation/, Build 36; accessed November 2008) to determine whether any CNVs in the general population were present in the 1p34.2p34.3 microdeletion region. No CNVs encompassed the entire deleted region, but we did find several smaller blocks of CNVs (figure 2A). The largest of these blocks, independently characterised by four groups (variations 3289, 4219, 5487 and 8322), was ∼435 kb in size and contained 11 genes (PPIE, PPT1, TRIT1, OXCT2, MYCL1, BMP8B, MFSD2, RLF, CAP1, TMCO2, and ZMPSTE24). Both copy number gains and losses of this region were reported in the general population.
Identification and characterisation of RIMS3 as a candidate gene for autism
We hypothesised that rare mutations in one or more genes in the 1p34.2p34.3 microdeletion region—excluding the 11 in the ∼435 kb polymorphic block—may be responsible for autism in other affected individuals as well. Interrogation of these ∼32 genes using published literature and online expression databases identified RIMS3 as the most promising candidate. Network analysis using Ingenuity Pathways Analysis software demonstrated that RIMS3 directly interacts with the protein tyrosine phosphatase PPFIA3 that belongs to the liprin-α gene family (supplementary figure 1). RIMS3 was also associated with many other autism candidate genes through an indirect interaction with FMR1, mutations of which cause fragile X syndrome (OMIM 300624).
To test the hypothesis that mutations in RIMS3 underlie the autism phenotype, we sequenced all RIMS3 coding regions, their associated splice sites, and the 5′ and 3′untranslated regions in 512 unrelated autism patients. We also sequenced these regions in 462 control subjects to capture the natural genetic diversity of RIMS3 in the general population. In the autism samples, we identified five novel coding variants (three non-synonymous and two synonymous) that were absent in control subjects and not reported in public DNA databases (Ensembl, http://www.ensembl.org/ and dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/, both accessed February 2009) (table 1). Demographic data for these five subjects, including sex, race, and ethnicity, are provided in table 1. We also identified a 5′untranslated region variant as well as one intronic variant located 30 bp upstream of the third exon. We found five different sequence changes in the controls (table 1).
RIMS3 mutation screening identifies putative mutations associated with autism
Sequencing of parental DNA indicated that all five coding variants were inherited. We performed segregation analysis in families by sequencing each variant in affected and/or unaffected siblings. In two multiplex families (AU0247 and AU0125, figure 3), the variant segregated with the autism phenotype in siblings. In family AU0247, the maternally inherited p.E177A variant first identified in patient HI0515 was also present in a sibling with NQA, but absent in 1161 control subjects. We reviewed available phenotype data on the carrier mother, who had depression and symptoms of bipolar disorder and anxiety. She also had a repaired cleft palate and had received speech language therapy (the father had received speech language therapy as well). The heterozygous sib had a ventricular septal defect that closed spontaneously by 1 year and diastasis recti. CNV analysis on this family provided on the AGRE website ruled out known CNVs associated with autism, including microdeletion of 22q11.

{kind=link}
{kind=link}
{kind=link}
Mutation analysis of RIMS3 identifies two missense variants that segregate with autism and autism related phenotypes. (A) The pedigree for family AU0247 shows that the p.E177A missense variant is present in a patient with autism (AU0247-03), in a sibling with Not Quite Autism (NQA) (AU0247-04), and in the mother who presents with psychiatric symptoms. Chromatograms indicate the presence of the A/C substitution. The SIFT program predicted the p.E177A substitution to be deleterious. (B) The pedigree for family AU0125 shows that the p.M260V missense variant is present in two siblings with autism (AU0125-03 and AU0125-04) as well as in the father who presents with psychiatric symptoms, including obsessive–compulsive disorder (OCD). Chromatograms indicate the presence of the A/G substitution. The variant is not predicted to affect protein function.
The p.E177A substitution, which changes a glutamic acid (acidic and polar) to an alanine (neutral and nonpolar) residue, was predicted to be deleterious according to both the SIFT and SNAP programs. We determined the extent of conservation of the E177 residue by comparing it against the RIMS3 primary protein structure of other species (http://genome.ucsc.edu/, hg 18, accessed February 2009). E177 was conserved among all 18 eutherian mammals with data available, but differs from the presumed ancestral Q177 residue found in six species of fish.
In family AU0125, the p.M260V exon 6 variant identified in patient HI0686 was not predicted to affect protein function by any of the three in silico protein programs. The carrier father had obsessive–compulsive disorder, depression, anxiety, and attention deficit hyperactivity disorder. The presence of psychiatric disorders in parents of children with autism has been observed previously in several studies,32–34 consistent with the hypothesis of multiple genes with small additive effects in autism. By parental report, both children had compulsions and rituals, and difficulties with changes in routines and their environment. A summary of phenotype information for the affected siblings in families AU0247 and AU0125 is provided in table 2, with detailed phenotype data for all members of families AU0247 and AU0125 included in the supplementary materials. Phenotypic data for the other three families with coding variants (AU0783, AU0679, and AU0841) were not available.
Phenotype analysis
We also sequenced RIMS3 in the boy with the 3.3 Mb deletion to test the hypothesis that the microdeletion might unmask a recessive allele on the non-deleted chromosome. We did not identify any putative functional coding variants.
In control subjects, we identified four coding variants that were distinct from those identified in autism patients; one of these was predicted to affect protein function (table 1). Three of the four variants were located in exon 2, a region that did not contain any of the five variants reported in the autism subjects. The fourth variant, p.I162V, was located in the same coding region in exon 4 that harboured the p.E177A substitution. Interestingly, the subject harbouring this variant presented with symptoms of obsessive–compulsive disorder. Phenotypic data on the parents of the control subjects were not available.
Discussion
We report a boy with postnatal microcephaly, minor dysmorphic features, mental retardation and autism associated with a 3.3 Mb deletion involving ∼43 genes in chromosome 1p34.2p34.3. The deletion is large and de novo, leading us to hypothesise that it is responsible for his abnormal phenotype. However, the specific genes responsible for different features of the 1p34.2p34.3 deletion phenotype are unknown.
Only a few patients with interstitial deletions of 1p34.2p34.3 have been reported. The closest match is the recent report of a Dutch boy with a 4.1 Mb deletion that completely overlaps the deletion we report, with loss of an additional 14 genes including the glucose transporter gene SLC2A1.35 Overall, he has a more severe phenotype than our patient with severe mental retardation and hypotonia, ‘profound’ microcephaly and epilepsy, as well as heterotopia and ponto-cerebellar atrophy on brain imaging. The more severe phenotype is not surprising given the severe phenotype associated with loss of SLC2A1 alone (OMIM 606777). Three other patients have been reported with deletions involving this general region. One boy with a much larger ∼17 Mb visible deletion had severe developmental delay, poor growth, and microcephaly.36 Two sibs who inherited a juxtaposed inversion (inv 1p22.3p34.1) and deletion (1p34.1p34.3) of this region presented with behaviour disorders, although their intelligence and appearance were normal.37 A detailed genotype–phenotype analysis involving 1p34.2p34.3 in these patients is provided in the supplementary materials.
Based on the reported altered regulation of RIMS3 in autism24 (see below) and our pathway analysis, we hypothesise that mutations in the synaptic protein RIMS3 is the primary contributor to the autism phenotype in the 1p34.2p34.3 microdeletion patient. A large number of autism related genes are involved in synapse function, neuronal cell adhesion, or both.38 Several of these have been implicated in autism based on cytogenetic rearrangements, CNVs or rare mutations, including NLGN3 and NLGN4,13 NRXN1,10 14 15 SHANK3,16–18 and CNTNAP2.19–21 RIMS3 belongs to the RIM protein family that function as important components of the presynaptic machinery for synaptic vesicle fusion and neurotransmitter release.39 Expression analysis of RIMS3 in the rat demonstrated that it is exclusively expressed in the brain40 and protein studies indicated that RIMS3 was detected only in rostral brain regions but not in spinal cord, hindbrain, or midbrain.40 Overexpression of RIMS3 has been demonstrated to greatly facilitate Ca2+ triggered exocytosis, supporting a role for RIMS3 as a regulator of exocytosis in the synaptic membrane.40 One recent study demonstrated that expression of RIMS3 is dysregulated in lymphoblastoid cells from autism patients with either maternal duplications of 15q11q13 or fragile X syndrome.24 Others have reported increased expression of RIMS3 in schizophrenia,41 42 a neuropsychiatric disorder hypothesised to have shared genetic aetiology with autism.43 Thus, several lines of evidence converge to support the hypothesis that abnormal RIMS3 function due to chromosomal imbalances or rare mutations may underlie autism. However, we cannot exclude the possibility that other genes within or near the ∼ 3.3 Mb 1p34.2p34.3 deletion may contribute to autism.
To address whether RIMS3 point mutations underlie autism, we sequenced the complete RIMS3 coding region in 512 unrelated autism patients. In total, five coding variants were identified in autism patients that were absent in ∼460 controls. None of these variants were de novo and all were inherited with no bias between maternal versus paternal transmission. We used several in silico programs to assess the functional impact of these variants on protein function, and identified one variant (p.E177A) that was predicted to be deleterious. This variant segregated with the autism phenotype in two sibs, was not identified in 1161 controls, and is highly conserved among eutherian mammals.
The p.E177A substitution resides in a C2B domain that is known to bind to several proteins including α-liprins, SNAP-25, and voltage gated calcium channels.44 45 However, the critical residues for some of these interactions are downstream of p.E177 and this amino acid is not conserved among the RIM family of proteins.40 α-Liprins are known to interact directly with CASK, a calcium/calmodulin dependent serine protein kinase that belongs to the membrane associated guanylate kinase family. CASK regulates the trafficking, targeting and signalling of numerous presynaptic proteins including neurexins, APBA1 (Mint1) and ion channels,46 and is also implicated in brain development.47 Interestingly, a C2B domain is also found in Doc2-α (Double C2-like domains, α), a protein that belongs to the same superfamily as RIMS3. DOC2A is one of ∼24 genes located within the 16p11.2 microdeletion-duplication region that has recently been detected in ∼1% of patients with autism.8 11 12
Although the p.E177A variant was not found in a large number of controls and was predicted to be deleterious using SIFT and SNAP, further studies are needed to prove whether the p.E177A substitution is pathogenic or not, as protein simulation programs do not substitute for experimental confirmation.29 48 Only two of the three in silico methods we used predicted a deleterious effect from the p.E177A variant. This is not surprising, given that each program varies in methodology, including differences in classification approaches (eg, machine learning vs rule based systems) and input information (eg, sequence data, functional annotation, protein structure, solvent accessibility).29 31 We also identified one variant in a control subject that was predicted to be deleterious. Only limited phenotype data are available for this subject, so we cannot determine whether it is a benign or weakly pathogenic variant. Detailed phenotypic assessment of this subject would help distinguish between these possibilities.
In summary, our data demonstrate that heterozygous loss of one or more of the 43 genes located within a ∼3.3 Mb interval on chromosome 1p34.2p34.3 are associated with abnormal development, including mental retardation, postnatal microcephaly and autism. Furthermore, our data suggest but do not prove conclusively that RIMS3 is an autism causative or contributory gene. To our knowledge, this is the first study to suggest that mutations in a RIM protein may underlie autism or other neurodevelopmental disorders. The identification of putative mutations in RIMS3 is consistent with the identification of CNVs and intragenic mutations of other pre- and post-synaptic proteins in patients with autism, and lends further support to the hypothesis that abnormalities in synaptic development and function underlie autism in some (and perhaps many) patients. Functional studies of RIMS3 variants such as p.E177A should provide additional insight into the role of pre-synaptic proteins in autism. Finally, the rare association of microcephaly and autism suggests that other gene(s) in the region are associated with brain growth.
Acknowledgments
We thank the parents of the first patient for offering so much of their time and support, and disclose that his mother and the senior author (WBD) are related. We gratefully acknowledge the resources provided by the AGRE Consortium and participating AGRE families. Additional autism families and the control samples were acquired from the NIMH Center for Collaborative Genetic Studies on Mental Disorders. We also thank Elizabeth Berry-Kravis of Rush University Medical Center for performing fragile X testing in our deletion 1p3 proband. This work was supported in part by grants from the National Institute of Neurological Disorders and Strokes to SLC (R01-NS051812) and WBD (R01-NS050375), and the Roswell Park Cancer Institute Comprehensive Cancer Center Grant (P30 CA01605632) to NN. We are grateful to Autism Speaks for awarding Postdoctoral Fellowships to Ravinesh A Kumar and Camille W Brune. AGRE is a program of Autism Speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M Lajonchere (PI).
References
Supplementary materials
Web Only Data 47/2/81
Files in this Data Supplement:
Footnotes
Supplementary figures and tables are published online only at http://jmg.bmj.com/content/vol47/issue2
Funding NIMH, NINDS, Autism Speaks.
Competing interests None.
Ethics approval This study was conducted with the approval of the University of Chicago.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.