Article Text

Abstract
Background Silver–Russell syndrome (SRS) is characterised by intrauterine growth restriction, poor postnatal growth, relative macrocephaly, triangular face and asymmetry. Maternal uniparental disomy (mUPD) of chromosome 7 and hypomethylation of the imprinting control region (ICR) 1 on chromosome 11p15 are found in 5–10% and up to 60% of patients with SRS, respectively. As many features are non-specific, diagnosis of SRS remains difficult. Studies of patients in whom the molecular diagnosis is confirmed therefore provide valuable clinical information on the condition.
Methods A detailed, prospective study of 64 patients with mUPD7 (n=20) or ICR1 hypomethylation (n=44) was undertaken.
Results and conclusions The considerable overlap in clinical phenotype makes it difficult to distinguish these two molecular subgroups reliably. ICR1 hypomethylation was more likely to be scored as ‘classical’ SRS. Asymmetry, fifth finger clinodactyly and congenital anomalies were more commonly seen with ICR1 hypomethylation, whereas learning difficulties and referral for speech therapy were more likely with mUPD7. Myoclonus-dystonia has been reported previously in one mUPD7 patient. The authors report mild movement disorders in three further cases. No correlation was found between clinical severity and level of ICR1 hypomethylation. Use of assisted reproductive technology in association with ICR1 hypomethylation seems increased compared with the general population. ICR1 hypomethylation was also observed in affected siblings, although recurrence risk remains low in the majority of cases. Overall, a wide range of severity was observed, particularly with ICR1 hypomethylation. A low threshold for investigation of patients with features suggestive, but not typical, of SRS is therefore recommended.
- Silver Russell syndrome
- mUPD7
- 11p15 hypomethylation
- phenotype
- imprinting
- clinical genetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Silver–Russell syndrome (SRS) is characterised by intrauterine growth restriction (IUGR), poor postnatal growth, relative macrocephaly, triangular facial appearance, asymmetry of the face and/or limbs, and fifth finger clinodactyly.1–3 In 1999, Price et al4 studied a cohort of 50 patients with SRS and suggested criteria for diagnosis. However, the relatively non-specific features of SRS present a continuing challenge to clinical diagnosis and to definition of its true spectrum and natural history.
SRS is genetically heterogeneous. The first molecular abnormality identified in a significant proportion of patients was maternal uniparental disomy for chromosome 7 (mUPD7).5 Evidence suggests this is present in around 5–10% of cases.6 7 More recently, abnormalities of chromosome 11p15 have been described. Chromosome 11p15 contains imprinted genes implicated in fetal growth, controlled by two imprinting control regions (ICRs): the telomeric ICR1 regulates expression of IGF2 and H19; the centromeric ICR2 controls expression of CDKN1C, LIT1 (KCNQ10T1) and other genes. Disturbances of this region are associated with the overgrowth disorder Beckwith–Wiedemann syndrome (BWS). Identification of maternally derived chromosome11p15 duplications in growth-restricted individuals, some with features of SRS,8 9 led to further investigation of this region in SRS.
In 2005, Gicquel et al10 reported loss of paternal methylation at ICR1 in five out of nine patients with typical SRS features. This was associated with biallelic expression of H19 and downregulated expression of the growth promoter IGF2. Subsequent studies suggest that up to 60% of patients with SRS have ICR1 methylation defects, depending on the clinical criteria used for selection of cases.7 11 To date only one patient has been reported with a maternally derived duplication restricted to ICR2.12
Initial reports of a relatively mild phenotype in association with mUPD713 have been corroborated by more recent comparisons with patients with ICR1 hypomethylation.11 14 15 However, retrospective analysis of all patients with mUPD7 published to date concluded that mUPD7 could not be reliably differentiated from ICR1 hypomethylation on clinical grounds alone.16 Clinical studies of several patient cohorts with ICR1 hypomethylation11 14–19 show in the majority a typical SRS phenotype comprising IUGR, postnatal short stature, characteristic craniofacial features and, often, asymmetry.11 14 15 18 A minority of patients have less typical features.17 20 21 Correlation between the level of ICR1 hypomethylation and the severity of SRS phenotype has been reported in several studies,10 14 17 20 although the majority of these have relied on retrospective data, which may be limited and/or incomplete. Comparisons of patients with ICR1 hypomethylation and mUPD7 are restricted by the small numbers of patients in the latter group.
We report a prospective study of clinical features among 64 patients with a positive diagnosis of SRS: 20 with mUPD7 and 44 with 11p15 hypomethylation. Findings in patients with mUPD and 11p15 methylation abnormalities were compared, allowing features more characteristic of each group to be defined. We also sought evidence of correlation between the level of 11p15 hypomethylation and severity of clinical features.
Methods
Patients
Sixty-four patients with either mUPD7 or 11p15 hypomethylation were ascertained, following referral to diagnostic laboratories within the UK and the Netherlands for investigation of a possible diagnosis of SRS, growth restriction or asymmetry. Molecular reports were obtained for all patients confirming positive diagnosis. Patients and their parents attended an appointment during which clinical information was recorded on a standard proforma, and the patient was examined and measured. To preserve consistency in recording of data, all patients were seen by one or more of a small group of experienced clinical geneticists (EW, MVH, DL, SP, CT, KT, JMC) with a special interest in SRS. Subsequently, all data and clinical pictures were evaluated by one coordinating clinical geneticist (EW). Additional information was also obtained from the hospital notes. Informed consent was obtained from all patients and/or their parents, and the study was approved by the Trent Research Ethics Committee.
Molecular analysis
DNA methylation of KCNQ1OT1 and H19 was measured in the Academic Medical Centre, University of Amsterdam, the Netherlands (laboratory 1; 21 patients), and, in the UK, the Wessex Regional Genetics Laboratory (laboratory 2; 19 patients), West Midlands Genetics Laboratory (laboratory 3; four patients) and South West Thames Molecular Genetics Diagnostic Laboratory (laboratory 4; three patients).
Methylation-specific multiplex ligation-mediated PCR analysis was the first-line test in laboratories 3 and 4, and the second-line test in laboratories 1 and 2. The SALSA MLPA kit ME-030 (MRC Holland, Amsterdam, The Netherlands) was used according to the manufacturer's instructions.22 In laboratories 1, 2 and 4, the cut-off for diagnosis of an epigenetic anomaly was a peak height ratio of ≥1.3 at two or more adjacent probes, and in laboratory 3 the cut-off was ≥1.25. Among 75 normal controls, such a ratio was not observed at two or more adjacent probes.22
In laboratory 1, methylation-specific high-resolution melt analysis (HRM-A) was used as first-line testing, as described.23 Hypomethylation was determined by visual comparison of case and control samples, an abnormal peak shape being absent from 45 normal controls. In laboratory 2, methylation-specific PCR (MS-PCR) was used as the first-line test, as described.24 A peak height ratio of 1.3 was taken as evidence of hypomethylation, such a ratio not being seen among 120 normal controls. For both HRM-A and MS-PCR, genomic DNA was bisulfite-modified using the EZ and EZ Gold DNA modification kits (Zymo Research, Orange, California, USA) according to the manufacturer's instructions. In all laboratories, testing for uniparental disomy of chromosome 11 was performed by standard analysis of microsatellite markers including D11S2071, D11S4046, D11STH, D11S1318 and D11SHBB.
Testing for uniparental disomy of chromosome 7 was performed by microsatellite analysis in laboratories 1–4 (13 patients) and a further six NHS service laboratories in the UK (eight patients). In laboratories 1–4, a minimum of six markers were used, including at least two each on 7p and 7q. Exclusion of mUPD7 required evidence of biparental inheritance at two or more markers, whereas a positive diagnosis required evidence of uniquely maternal inheritance of two or more markers. In the remaining laboratories, data from diagnostic reports confirmed that a minimum of three microsatellite markers were tested, with positive diagnosis requiring evidence of uniquely maternal inheritance of at least two markers.
Clinical scoring
Patients were scored using the five key criteria (birth weight ≤−2 SD from mean; poor postnatal growth ≤−2 SD from mean; preservation of occipitofrontal circumference; classic facial features; asymmetry) suggested by Price et al.4 Patients in our study found to have at least four of these features were described as having ‘classical’ SRS. Limb asymmetry was scored as present if there was ≥1 cm arm and/or leg length difference; facial asymmetry was scored subjectively.
Laboratory staff were blind to the clinical scoring/phenotype of the patients tested. Scoring was carried out by one of the investigators not involved in examination of the patients and also blind to the methylation index results (SK).
Statistical analysis
Statistical comparisons between the two molecular subgroups were made using the Fisher exact test, the unpaired t test and the Mann–Whitney test, as appropriate. Correlation of methylation index with a number of clinical parameters was analysed using Pearson correlation, Spearman's rank correlation and the unpaired t test, as appropriate. Statistical significance was set at ≤0.05.
Results
A total of 64 patients were included in this study: 44 with11p15 methylation abnormalities and 20 with mUPD7. The mean age in the two groups was similar: 6.3 years (range 0.8–26.8) for ICR1 hypomethylation; 7.3 years (range 1.3–17.9) for mUPD7.
The majority of patients with abnormal 11p15 methylation had abnormalities restricted to ICR1. One patient reported on previously was known to have mosaic mUPD11,25 and two more had maternal duplication of ICR1 and ICR2 (see supplementary text and figure 3 online). All three of these patients have typical SRS features.
In one family an affected brother and sister were found to have ICR1 hypomethylation. Their parents and two other unaffected siblings had normal methylation patterns.
Clinical score
Of the patients with ICR1 hypomethylation, 61% had at least four of the five key features, compared with just 20% of patients with mUPD7 (p=0.003). The mean scores for patients with ICR1 hypomethylation and mUPD7 were 3.7 and 3.0, respectively. However, the greatest range in severity was associated with ICR1 hypomethylation (range 2–5, compared with 2–4 for mUPD7). Four (9%) patients with ICR1 hypomethylation and five (25%) with mUPD7 had just one or two of these features. Of these, four were referred for testing on the basis of asymmetry (hemihypotrophy) and five due to prenatal and/or postnatal growth failure.
Two patients with normal growth parameters were referred for investigation of asymmetry. One patient with mUPD7 had typical facial features, but birth weight −1.53 SD and height −1.3 SD at 7 years. Another patient with 11p15 hypomethylation had fifth finger clinodactyly, but no other features of SRS, with birth weight −1.33 SD and height +0.2 SD at 10 months.
The frequency of clinical features found in the two molecular subgroups is summarised in table 1.
Clinical features in patients with Silver–Russell syndrome with hypomethylation of the imprinting control region (ICR) 1 and maternal uniparental disomy of chromosome 7 (mUPD7)
Growth parameters
From maternal recollection, IUGR was suspected in 89% cases with ICR1 hypomethylation and 70% with mUPD7 (p=0.08). In both groups, the average gestation at which IUGR was detected was 23 weeks, probably reflecting the fact that most women have anomaly scans at around this stage of pregnancy.
Overall, 78% of patients had a birth weight ≤−2SDS with a wide range, particularly among patients with ICR1 hypomethylation (range −4.88 to −0.5 SD, mean −2.49 SD). Patients with mUPD7 had a mean birth weight of −2.24 SD (range −3.29 to −1.29 SD). Birth weight was therefore more frequently ≤−2SDS with ICR1 hypomethylation, whereas height at examination was more frequently ≤−2 SDS with mUPD7.
In total, 30% of patients had received growth hormone. Insufficient retrospective data were available to analyse the difference in the effect of growth hormone treatment. In patients not treated with growth hormone, those with ICR1 hypomethylation (53%) were less likely to show postnatal reduction in height SDS than those with mUPD7 (78%) (p=0.26). Eight patients had reached their final height, five of which had been treated with growth hormone (see supplementary table online). Numbers were too small to allow meaningful analysis of the effect of growth hormone on final height.
Developmental delay
Global developmental delay was described in 34% cases. Severe delay was uncommon, being described in only two children, both with ICR1 hypomethylation. One was diagnosed following investigation for asymmetry. However, he was felt to be very atypical for SRS, his growth parameters were normal, and further investigation is ongoing to look for an additional cause of his problems. The other had suffered a cardiac arrest at 9 months following repair of a ventricular septal defect. If these two children are excluded from the analysis, moderate delay was seen in one (2%) patient with ICR1 hypomethylation and two (10%) with mUPD7. In the remainder, developmental problems were mild. In those patients without global delay, gross motor delay was still common, with mean age at walking of ∼20 months in both groups.
Behavioural problems were uncommon and mild. Three children (one with mUPD7 and two with ICR1 hypomethylation) were reported by their parents as being hyperactive. Only one child had been referred for further assessment for behavioural problems.
Congenital anomalies
Major congenital abnormalities were markedly more common in those patients with ICR1 hypomethylation (table 2). Camptodactyly was seen in 19% overall and appears to gradually progress in severity with age. Restriction of movement in other joints was confined to patients with ICR1 hypomethylation. Only upper limbs were affected, with four patients having limited elbow extension bilaterally/unilaterally and one having bilateral reduction of shoulder movements.
Congenital anomalies in patients with Silver–Russell syndrome with hypomethylation of the imprinting control region (ICR) 1 and maternal uniparental disomy of chromosome 7 (mUPD7)
Hypoglycaemia
Excessive sweating was reported by 67% of parents. This may have represented hypoglycaemia, but in most cases had not been formally investigated. Since there was no significant difference in the frequency of documented evidence of hypoglycaemia in either group, it is unlikely to account for the difference in rate of global developmental delay.
Feeding difficulties
Feeding difficulties were scored according to severity (1=normal, 2=mild, 3=frequent/long feeds, 4=prolonged nasogastric feeds, 5=gastrostomy insertion). The average score for mUPD7 was slightly higher than that for ICR1 hypomethylation (mean (SD) 4.7 (1.3) and 3.4 (1.4), respectively), although this did not reach statistical significance (p=0.48).
Movement disorders
Intriguingly, one patient with mUPD7, aged 14.9 years, was described as having intermittent episodes of head shaking. Another patient, aged 14.2 years, has a slight tremor affecting his left arm. One further patient with mUPD7 had myoclonic jerks in infancy (from 3 weeks to 1 year), which had subsequently resolved. No patients with ICR1 hypomethylation had similar problems.
Facial features
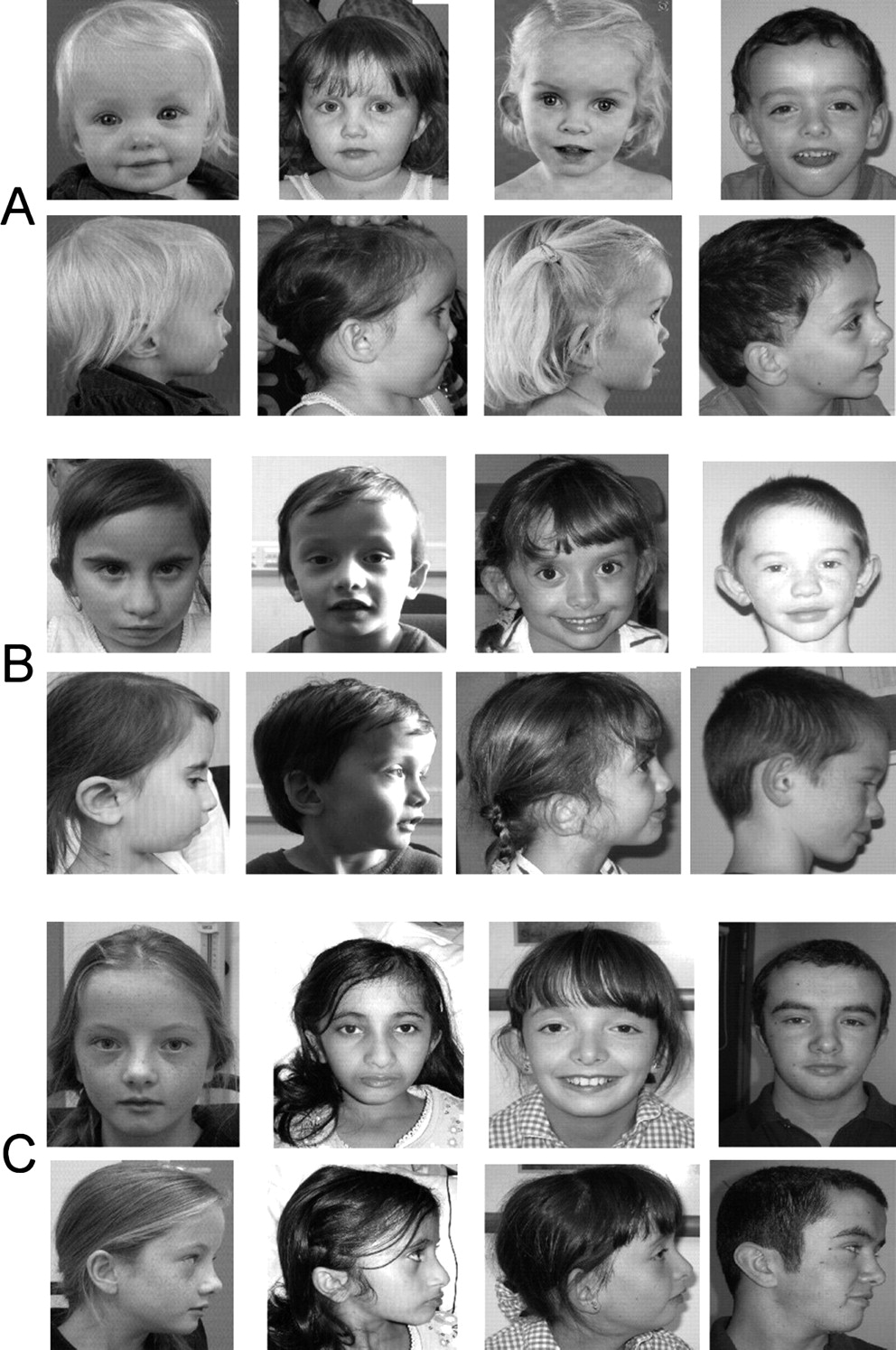
Figures 1 and 2 show facial features for many of the patients with ICR1 hypomethylation and mUPD7 included in this study. The photographs illustrate how facial features become less striking with age. Owing to difficulties obtaining early pictures and growth data from many of the older patients, clinical features such as frontal bossing, triangular face and micrognathia were scored according to the appearance of the patient at examination (table 1). In addition, data were analysed for triangular facies and frontal bossing in those patients under 5 years at examination. As expected, this showed increased frequency of specific features at this age (triangular face in 67% of ICR1 hypomethlyation and 100% of mUPD7; frontal bossing in 81% of ICR1 hypomethylation and 67% of mUPD7).

Facial appearance of patients with hypomethylation of the imprinting control region (ICR) 1 at different ages. Group A: 1.4–3.3 years; group B: 3.3–4.7 years; group C: 5.9–9.5 years; group D: 9.8–26.6 years.
{kind=link}
{kind=link}
Facial appearance of patients with maternal uniparental disomy of chromosome 7 with increasing age. Group A: 1.3–3.8 years; group B: 4.0–7.9 years; group C: 10.0–14.2 years.
Correlation with methylation index
Data on methylation index was available for 29 of the 44 patients with ICR1 hypomethylation. Clinical score, birth weight SDS, postnatal height SDS, severity of feeding difficulties, and the presence of developmental delay, asymmetry and/or congenital anomalies were all analysed for correlation with level of ICR1 hypomethylation. No evidence was found for correlation between the level of ICR1 hypomethylation and clinical severity (table 3).
Correlation of level of hypomethylation of the imprinting control region (ICR) 1 with clinical severity
Assisted reproductive technology (ART)
All five cases conceived as a result of in vitro fertilisation treatment, including one via ovum donation and one via intracytoplasmic sperm injection, were in the ICR1 hypomethylation group. However, this difference between the groups did not reach statistical significance.
Discussion
Clinical diagnosis of SRS remains difficult, as many of its features are non-specific, diagnostic criteria are not universally agreed, and features are most striking in early childhood, making assessment of older patients difficult. By collecting clinical data from patients with a positive molecular diagnosis of SRS, we were able to include patients irrespective of whether their referring physician felt them clinically ‘typical’. This reduced reporting bias. In contrast with previously published studies, our data were gathered prospectively by a small number of experienced geneticists, allowing both detailed and consistent analysis of the phenotype.
Several scoring systems for clinical diagnosis of SRS have been proposed.4 11 15 Most recently, Bartholdi et al15 developed criteria to score 168 patients with suspected SRS, which were fulfilled by all patients found to have ICR1 hypomethylation, but only seven of 10 patients with mUPD7. These criteria could not be applied in our study because data such as birth occipitofrontal circumference and length were often unavailable, and the early age of many participants precluded scoring for normal cognitive development (defined as attending regular school). We therefore used the criteria suggested by Price et al,4 as complete data were available for scoring in all patients. However, we recognise that these criteria do not include feeding difficulties, which are a major feature of this condition.
Clinical features in mUPD7 and ICR1 hypomethylation
Our study included sufficient patients with mUPD7 to allow statistical comparison of features with ICR1 hypomethylation cases. Consistent with other studies,11 14 15 17–19 we found that 61% of patients with ICR1 hypomethylation had ‘classic’ features of SRS, compared with only 20% of patients with mUPD7.
Patients with ICR1 hypomethylation were less likely to show postnatal reduction in height SDS than those with mUPD7, although numbers were too small to reach statistical significance. Binder et al19 showed that children with mUPD7 have a significantly higher birth length but lose height SDS post partum, whereas those with 11p15 hypomethylation show no change in height SDS. The same study noted a trend towards more height gain with growth hormone therapy in children with mUPD7 than those with ICR1 hypomethylation.19 In our study, insufficient data were available to test this observation. However, two adults with ICR1 hypomethylation who had been treated with growth hormone achieved final heights well within the normal range. The possible differential effect of growth hormone in these two subgroups is an important clinical question which deserves further detailed and prospective investigation.
Asymmetry was significantly more common with ICR1 hypomethylation. This finding is in keeping with previous reports4 13 15 16 and may reflect mosaicism for hypomethylation at the tissue level.
We found psychomotor retardation in approximately one-third of patients with SRS, in keeping with previous observations.26 Patients with mUPD7 were more likely to have delayed development and to have a statement of education. Global delay was mostly mild, although moderate delay was more common with mUPD7. Speech delay and referral for speech therapy were more common with mUPD7, as reported in previous studies.13 This finding has been linked to the absence of paternal FOXP2 expression, as seen in other patients with developmental verbal dyspraxia.27 Early motor delay was also relatively common in both groups and may result from a combined effect of low muscle bulk and relatively large head size in infancy.
Feeding difficulties are well recognised as a major feature of SRS.28 Parents of children from both groups in this study often commented on their lack of interest in sucking and absence of hunger from birth.
In the SRS cohort of Price et al,4 22% had generalised camptodactyly. It has been hypothesised that this is specific to patients with ICR1 hypomethylation.17 Bruce et al14 reported on several patients with ICR1 defects and joint contractures (including limited elbow extension) and/or other skeletal abnormalities. In our cohort, limited elbow extension was seen in four patients. Interestingly, one patient was found to have radial hypoplasia. Although this feature is not usually associated with SRS, at least one other patient with ICR1 hypomethylation has been described with thumb hypoplasia.14
In common with other studies,14 major congenital anomalies were significantly more common in patients with 11p15 involvement. Overall, major congenital anomalies appear to be much more suggestive of, though not exclusive to, ICR1 hypomethylation.
Importantly, we observed two teenage patients with mUPD7 and mild movement disorders; a further patient had a history of myoclonic jerks in infancy. This is particularly interesting in light of a recent report of myoclonus-dystonia in a patient with mUPD7.29 Myoclonus-dystonia typically presents before adulthood with mild dystonia (such as cervical dystonia or writer's cramp) and/or myoclonic jerks. The disorder is associated with paternally derived mutations in the imprinted ε-sarcoglycan (SGCE) gene on chromosome 7q21. It may not previously have been noted with mUPD7, as symptoms are relatively mild and may start in later childhood. In addition, other genes may modify the development of this condition, as has been noted in dystonia due to DYT1 and DYT6 mutations.30
The ‘classical’ facial features of SRS (triangular-shaped face, frontal bossing, down-turned corners of the mouth, and micrognathia) were apparent in many but not all patients in both groups. One retrospective study suggested a higher frequency of macrocephaly and frontal bossing in patients with ICR1 hypomethylation, and a higher incidence of triangular facies with mUPD7.16 These previous observations are supported by our analysis of facial features in patients under 5 years.
It is well recognised that patients with BWS due to uniparental disomy and imprinting defects of ICR1 are at increased risk of embryonal tumours.31 However, to date none of the SRS patients with hypomethylation of H19 reported in this study have developed tumours. Taken together with data from previous studies,11 14 15 17 20 there is currently no evidence for an increased childhood tumour risk in patients with ICR1 hypomethylation.
Variation in severity of features
While most studies have found no evidence for ICR1 hypomethylation in cohorts of patients with isolated prenatal or postnatal growth retardation,11 14 15 18 incomplete SRS phenotype has been described with ICR1 methylation abnormality.17 20 21 Patients presenting primarily with hemihypotrophy or with mild prenatal and/or postnatal growth failure therefore form part of the spectrum of ICR1 hypomethylation.
The range of phenotypic severity has been linked to the level of ICR1 hypomethylation. Bruce et al14 reported correlation of severe ICR1 hypomethylation with the presence of asymmetry, micrognathia and congenital anomalies. It was suggested that 35% of the variation in clinical severity could be explained by the level of H19 hypomethylation. However, we found no statistical correlation between methylation index and degree of clinical severity in 29 patients studied.
Other factors may also influence the degree to which patients with ICR1 hypomethylation are affected. Methylation status in all our patients was analysed in DNA from blood lymphocytes, but clinical severity may reflect tissue-level mosaicism. Furthermore, Bartholdi et al15 described selective hypomethylation of either H19 or IGF2 in 11/42 and 3/42 patients, respectively, associated with amelioration of phenotype. We had insufficient molecular data to perform a similar analysis in this study. Another potential explanation for the clinical variation may be hypomethylation of multiple imprinted loci in some patients.32 Fourteen patients in this study were investigated for hypomethylation of multiple imprinted loci, three of whom showed additional methylation anomalies; the significance of this remains to be determined.
Assisted reproductive technology
Several recent studies have reported an increased frequency of ART conceptions in children with BWS or Angelman syndrome.33 These findings are consistent with reports of imprinting defects in animal studies after in vivo embryo culture. Evidence also exists for an increased frequency of ART in association with SRS.34 The general rate of ART is 1–3%, of which low birth weight is a recognised complication.35 In this study, all five patients conceived as a result of ART had ICR1 hypomethylation (11% of this group), supporting an association between ART and imprinting defects in SRS. There is evidence that the increased incidence of BWS and Angelman syndrome after ART is associated with fertility problems,36 but patients with SRS were not included in this study. Interestingly, the reported rate of infertility (taking over 1 year to conceive) in our two subgroups was similar (18% with ICR hypomethylation and 20% with mUPD7). This suggests that ART may also have a direct effect. However, much larger and more rigorous studies are required to investigate this further.
Familial recurrence
The vast majority of cases of SRS are non-familial, but a few exceptions are reported in the literature, notably those described recently by Bartholdi et al.14 One recurrence within affected siblings was found in the present study. Microsatellite analysis in the family was inconsistent with an imprinting centre defect within 11p15. In another study, methylation analysis in sperm from a patient with 11p15 hypomethylation showed a normal sperm-specific methylation pattern with full methylation at two loci within this region.19 These findings suggest that epimutation is reversed in the male germline. An alternative explanation for recurrence, consistent with these findings, is an inherited alteration of a trans-acting factor responsible for maintenance of the imprint after fertilisation.
Adult patients
Only eight of our cohort had reached postpubertal age, possibly reflecting several factors: molecular diagnosis of SRS has only recently become widely available; SRS cases may become lost to follow-up; and the characteristic features become less noticeable with time. It has been hypothesised by Barker and Hales37 that the epidemiological association between poor fetal and infant growth and the subsequent development of type 2 diabetes results from the effects of poor nutrition in early life, which in turn produces permanent changes in glucose/insulin metabolism. However, there is, as yet, no evidence to suggest that patients with SRS are at increased risk of developing type 2 diabetes or other metabolic problems in adulthood. In this study, detailed endocrine work-up had only been carried out in one adult patient (at 26 years). Longer-term, systematic endocrine follow-up, looking for evidence of metabolic abnormalities in patients with mUPD7, ICR1 hypomethylation and idiopathic SRS will be important.
Conclusions
This study is restricted to SRS patients with known molecular abnormalities. In ∼30% of patients with a clinical diagnosis of SRS, the underlying molecular defect is unknown. Ideally, molecular karyotype analysis should be carried out in patients where the diagnosis of SRS is considered but mUPD7 and ICR1 hypomethylation have been excluded. Recent studies have shown that a small proportion of such patients have cryptic chromosome rearrangements.38 However, they are often labelled as ‘mild’ SRS, and this reinforces the importance of careful clinical assessment to try to reduce the heterogeneity of patients with idiopathic SRS.
On the other hand, application of a strict scoring system risks missing patients with either mUPD7 or a mild phenotype associated with ICR1 hypomethylation. Only 61% of hypomethylation 11p15 and 20% of mUPD7 cases in our study would have been diagnosed as ‘classical’ SRS according to the criteria of Price et al.4 No single clinical feature was present in all cases and even low birth weight in only 78% overall. We would therefore recommend a low threshold for investigation of SRS, and, as it is difficult to differentiate between mUPD7 and ICR1 hypomethylation on the basis of clinical features alone, testing for both should be carried out.
Acknowledgments
We thank all the parents and children in the UK and the Netherlands who have participated in the study. We are grateful to the many clinicians who have helped recruit patients to the study and provided clinical data. Hillary Bullman and Margaret Lever from Wessex Regional Genetics laboratory tested many of the samples from patients in UK and provided useful feedback on the paper. We also thank Paul Bassett for his clear statistical advice.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Funding Child Growth Foundation, 2 Mayfield Avenue, Chiswick, London W4 1PW.
Competing interests None.
Patient consent Informed consent was obtained from all patients/parents for publication of photographs.
Ethics approval This study was conducted with the approval of the Trent Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.