Article Text

Abstract
Background Cone-rod dystrophy is a retinal dystrophy with early loss of cone photoreceptors and a parallel or subsequent loss of rod photoreceptors. It may be syndromic, but most forms are non-syndromic with autosomal dominant, autosomal recessive or X-linked recessive inheritance.
Methods and results We identified a small consanguineous family with six patients with cone-rod dystrophy from the Faroe Islands. Homozygosity mapping revealed a single homozygous locus of 4.2 Mb on chromosome 10q23.1–q23.2, encompassing 11 genes. All patients were homozygous for a 1-bp duplication in PCDH21, c.524dupA, which results in a frameshift and a premature stop codon (p.Q175QfsX47).
Conclusion To our knowledge, this is the first report of mutations in PCDH21 as a cause of human disease. PCDH21 is highly expressed in the retinal photoreceptor cells. It encodes protocadherin 21, which belongs to the cadherin superfamily of large cell surface proteins characterised by a variable number of extracellular cadherin domains. A PCDH21 knockout mouse model has previously shown loss of photoreceptor cells and abnormal cone and rod function, similar to the findings in the patients.
- Clinical genetics
- ophthalmology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Cone-rod dystrophy (CORD (MIM 120970)) is a group of inherited disorders that belong to the generalised retinal dystrophies. The prevalence of CORD is 1/40 000. CORD is characterised by early loss of cone receptors accompanied by loss of rod receptors, in contrast to retinitis pigmentosa (RP), where the loss of cone photoreceptors occurs secondarily to rod degeneration. The loss of cone photoreceptor cells leads to visual loss, visual field loss, abnormalities of colour vision and variable degrees of photophobia and nystagmus. The loss of rod function leads to night blindness. The onset is usually in late childhood or early adulthood. Fundus examination shows focal macular retinal pigment epithelial (RPE) atrophy or a bull's eye maculopathy in the early stages, and in the later stages peripheral RPE atrophy, retinal hyperpigmentation, arteriolar attenuation and optic disc pallor occurs. Absent or severely impaired cone function is found on electroretinography (ERG) and psychophysical testing.
CORD may be syndromic, for example, in Alstrom syndrome and spinocerebellar ataxia type 7,1 2 but most patients have non-syndromic CORD. Several genes have been identified in non-syndromic CORD: CRX, GUCY2D, RIM1, RDS, GUCA1A and AIPL1 in autosomal dominant CORD, RPGR in X-linked recessive CORD, and ABCA4, RPGRIP1, CNGA3 and ADAM9 in autosomal recessive CORD.3 4 In addition, four loci are known, where the disease-causing gene has not been yet been identified.
We have identified a small consanguineous family with CORD from the Faroe Islands, which is a small archipelago in the North Atlantic Ocean between Iceland and Norway. The present population was founded around 825 AD by migration from the Western part of Norway8 and remained small and isolated for a long time, with an estimated size of 4000 in the fourteenth century and 9000 in the nineteenth century. During the last 150 years, the population has expanded to the present size of around 50 000.9 No major bottlenecks are thought to have occurred.5 A founder effect is responsible for a high incidence of some disorders in the Faroe Islands, for example, cystic fibrosis6 and mitochondrial encephalomyopathy with elevated methylmalonic acid.7
Methods
Human participants
The consanguineous family consists of three men and three women affected with CORD (figure 1A). Genealogical studies revealed a common ancestor six generations back, born in 1687, of the parents of patients II:4, II:6 and II:10, and the mutation carrier III:1. The Faroese population has been studied extensively for hereditary ophthalmological disorders, but to our knowledge, no other Faroese persons have a diagnosis of CORD.
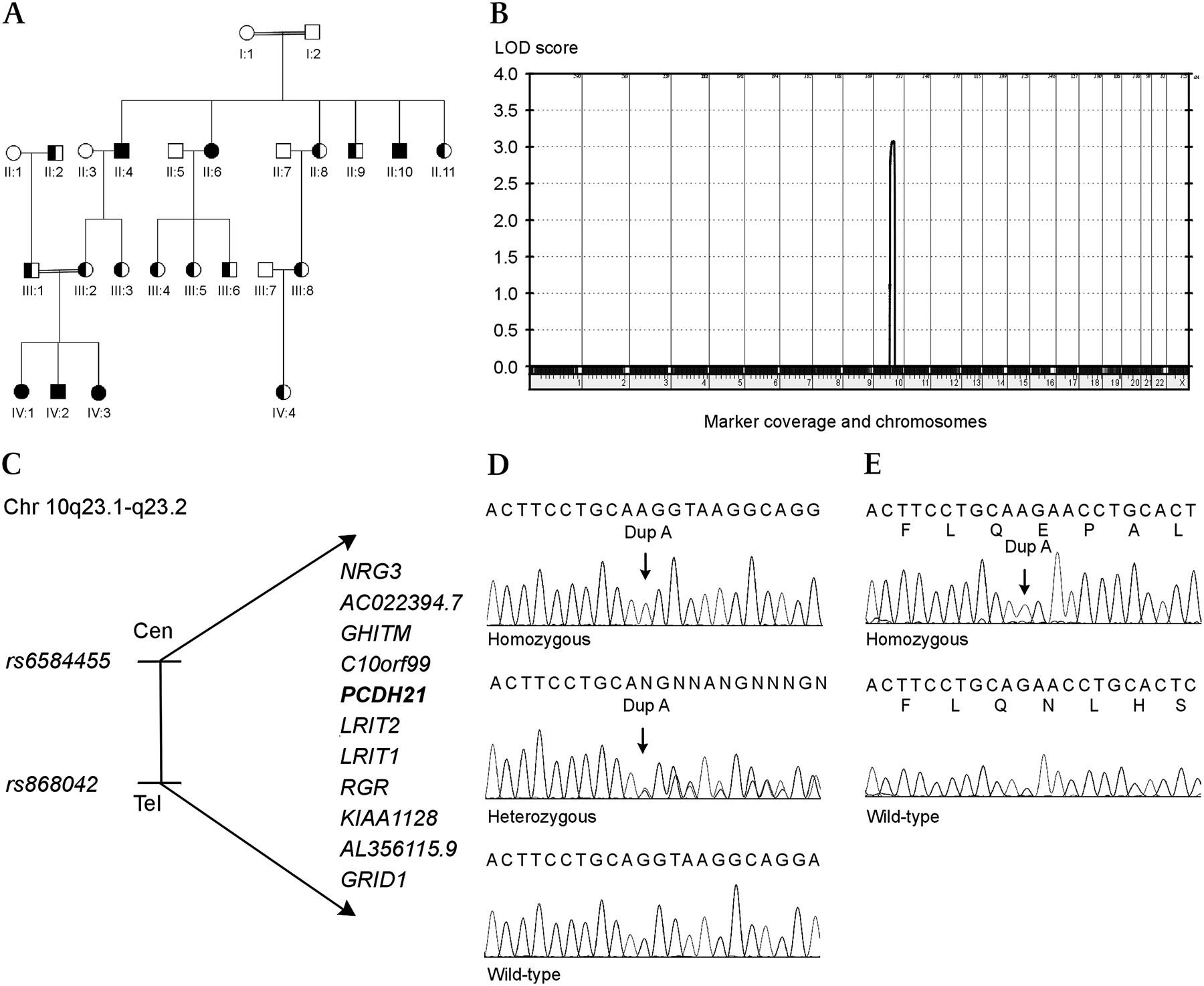
Pedigree of the family with CORD, linkage analysis, genes in the candidate region and mutation analysis of PCDH21. (A) Pedigree of the consanguineous family with a PCDH21 mutation. Filled symbols are homozygous affected individuals, half-filled symbols are heterozygous carriers. (B) Results of the parametric multipoint linkage analysis using Genehunter software, which shows a single significant peak on chromosome 10, with a LOD score of 3.1. (C) The 11 genes in the candidate region on chromosome 10q23.1–23.2. (D) Mutation analysis of PCDH21 shows the homozygous c.524dupA mutation (left), a heterozygous carrier (middle) and the wild-type (right). (E) Analysis of cDNA from fibroblasts from a homozygous patient shows that the mutation results in a frameshift (top). Wild-type is shown for comparison (bottom).
The study complied with the ethical guidelines of the Declaration of Helsinki. It was approved by the Faroese Ethical Committee and carried out in collaboration with the governmental Genetics Resource Centre of the Faroe Islands. The family gave informed consent for the study.
The pedigree analysis provided strong evidence of autosomal recessive mode of inheritance, and all affected persons were assumed homozygous for a mutant allele.
DNA and RNA extraction and cDNA synthesis
DNA was extracted from peripheral blood using standard procedures. RNA was extracted from cultured fibroblasts with the RNAeasy Mini kit (Qiagen, Inc., Valencia, California, USA) and reverse transcribed to cDNA with the SuperScript II Reverse Transcriptase kit (Invitrogen, Carlsbad, California, USA).
Microarray analysis
DNA from the six affected patients (II:4, II:6, II:10, IV:1, IV:2 and IV:3) was used for a genome-wide search for homozygosity with the Affymetrix GeneChip 50K Xba array (Affymetrix Inc., Santa Clara, California, USA). In brief, 250 ng of DNA was digested with the restriction enzyme XbaI, mixed with Xba adapters and ligated with the T4 DNA ligase. The ligated DNA was PCR-amplified in four PCRs, pooled and purified. The purified PCR product was fragmented with DNase I and end-labelled with biotin. The samples were hybridised to an array for 18 h in a hybridisation oven. The array was washed, stained and scanned with an Affymetrix GeneChip scanner 3000. Affymetrix software was used to analyse the data, which were exported to an Excel file.
Linkage analysis
Multipoint parametric linkage analysis was performed with the GeneHunter program on the EasyLINKAGE V.5.08 plus platform.10 An autosomal recessive mode of inheritance with complete penetrance was assumed, and the disease allele frequency was set at 0.001.
Mutation analysis
PCR was performed with the Promega kit and the following conditions: 0.2 mM dNTPs, 1× buffer, 1.5 mM MgCl2, 0.5 mM of each primer, 10 ng template and 1.5 U polymerase in a total volume of 50 μl. The PCR program was 94 C for 2 min, 35 cycles of denaturing at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 30 s and a final extension step of 72°C for 7 min. Primer sequences are available on request.
Sequencing was performed with ABI Big Dye Terminator v. 1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, California, USA). The sequencing reactions were vacuum-purified with the Montage Seq96 Sequencing Reaction Cleanup Kit (Millipore SA, SaintQuentin, France) and analysed on an ABI 3130xl Gene Analyzer (Applied Biosystems).
The data were analysed with Sequencing Analysis Software V.5.2 from Applied Biosystems, and mutation screening was performed using Mutation Surveyor V.3.1 Software (SoftGenetics).
Results
Clinical findings
The three oldest affected siblings (patients II:4, II:6 and II:10) reported decreased vision from around age 17 years. In addition to CORD, two of the patients (patients IV:1 and IV:3) had oculocutaneous albinism, which caused nystagmus and reduced visual acuity from early infancy. The genetic background for their albinism is unknown, but no mutations were found by sequencing TYR and OCA2. Patient IV:2 denied visual difficulty in daylight when examined at the age of 15. He had, however, always had difficulties with nighttime orientation. Patient II:10 complained of impaired vision at night and of delayed adaptation to sudden changes in light intensity.
Visual acuity varied between 0.67 and 0.02 (table 1). In patient II:4, large inter-ocular differences in visual acuity were observed due to a large scar in the left macula, which was likely caused by a foetal toxoplasmosis infection. The inter-ocular difference in visual acuity in patient II:10 was explained by a left-sided squint in childhood.
Clinical and electrophysiological characteristics of patients with CORD caused by PCDH21 mutations
Retinal examination showed both central and peripheral pigment abnormalities (figure 2). The foveal changes consisted of irregular pigmentation and atrophy of the RPE with a horizontal oval-shaped or ill-defined “moth-eaten” appearance. In patient IV:2, the foveal regions had an extensive sheen. In the two patients with oculocutaneous albinism (patients IV:1 and IV:3), the changes in the foveal regions were less obvious due to the scarcity of pigment. The peripheral pigment disturbances included diffuse RPE atrophy and black hyperpigmentations of polymorphic shape, some of which were filiform and sheeted the vessels. The amount of pigmentation varied considerably among patients of the same age but showed some correlation to the extent of visual field constriction. In the oldest patients, the optic discs looked moderately atrophic, and the retinal vessels were universally constricted.

Fundus pictures of the right eye. (A) Diffuse atrophy of the retinal pigment epithelium (RPE) and extensive sheen in the macular region (patient II:4). (B) Numerous, polymorphic, intraretinal hyperpigmentations in the upper temporal midperiphery (patient II:4). (C) Irregular foveal degeneration and normal arteriolar caliber (patient II:6). (D) Irregular RPE atrophy in the macular region, attenuated arterioles and paleness of the optic nerve head (patient II:10). (E) Albinotic fundus masking the macular changes (patient IV:1). (F) Foveal atrophy and attenuated arterioles (patient IV:2).
Visual field measurements with a large object showed considerable variation; patient II:4 had only narrow peripheral remnants left in the lower visual fields and a paracentral preserved island of 10°, whereas patients II:6 and II:10 had normal or slightly constricted outer field limits. In patient II:6, a central scotoma measuring 5° was found. The smallest test objects revealed concentric constriction in patients IV:1 and IV:2. Colour vision was severely impaired in the two patients tested (patients II:6 and II:10 at ages 36 and 38 years, respectively). Dark adaptation was tested in two patients (patients II:10 and IV:2 at ages 38 and 15 years, respectively). It showed a monophasic and protracted course, with only a slight elevation of the final threshold of 0.5–1 log unit in patient II:10 (figure 3).

{kind=link}
{kind=link}
{kind=link}
Dark adaptation in patients with CORD due to PCDH21 mutations. The dark adaptation curves in patients II:10 and IV:2 show a protracted initial phase and an absent mesopic break, resulting in a monophasic, straightened course compared to the normal diphasic course.
The ERG of patient II:10 showed severely reduced amplitudes and prolonged photopic implicit times (table 1). Patient IV:1 had normal amplitudes at the age of 2 years but prolonged cone implicit times. Eight years later, the ERG showed absent response to low-intensity stimuli and a 50% reduction in the response to high-intensity flash. In patient IV:2, the ERG response was absent at age 17 years.
Patients II:6 and II:10 reported normal smell sense.
Mapping of a gene for CORD to chromosome 10
A frequent cause of CORD is mutations in ABCA4, and we, therefore, analysed the SNP data for linkage to ABCA4. Assuming autosomal recessive inheritance and a homozygous or compound heterozygous mutation, we did not find evidence of linkage to ABCA4 (see online supplementary table 1).
Genome-wide analysis of SNP data for homozygosity revealed a single homozygous region on chromosome 10q23.1–q23.2 (see online supplementary table 2); a significant LOD score of 3.1 was found by multipoint linkage analysis (figure 1B). The linkage interval had a size of 4.2 Mb and was flanked by the heterozygous markers rs6584455 and rs868042 (figure 1C).
The region encompasses 11 genes, of which three were candidate genes: RGR, which had previously been shown to cause autosomal recessive retinitis pigmentosa in humans, LRIT1, which is highly expressed in retina, and PCDH21, where a knockout mouse model had shown loss of cone and rod photoreceptor cells.11–13
Screening of candidate genes
No mutations were found in RGR or LRIT1. Sequencing of PCDH21 revealed a 1 bp insertion, c.524dupA (figure 1D). The six patients were homozygous for the mutation, and 12 family members were heterozygous carriers (figure 1A). Individuals II:1 and II:3 did not carry the mutation; no DNA was available from the remaining family members.
The 1 bp duplication is expected to cause a frameshift resulting in a premature stop codon (p.Q175QfsX47), which was confirmed by analysis of cDNA from fibroblasts (figure 1E).
Screening of 159 anonymous Faroese controls for the mutation revealed three heterozygous carriers, corresponding to a carrier frequency of 1.9%. None of the controls were homozygous for the mutation.
Discussion
To our knowledge, this is the first report of mutations in PCDH21 as a cause of human disease. In a previous study, two heterozygous missense mutations were found in three patients with autosomal recessive RP, but a second mutation was not found; hence, the pathogenicity of the mutations is unclear.14
The PCDH21 mutation was identified in a small consanguineous family from the Faroe Islands, and a carrier frequency of 1:53 (1.9%) was found. A high carrier frequency is found in other disorders in the Faroe Islands that are caused by founder mutations, for example cystic fibrosis (1:24) and mitochondrial encephalomyopathy with elevated methylmalonic acid (1:33).8 9
PCDH21 was identified by Rattner et al by subtractive hybridisation and high-throughput screening in a search for genes specifically expressed in the retina.13 At the same time, Nakajima et al identified PCDH21 in a search for genes that encode proteins with cadherin domains.15 PCDH21 is conserved among different vertebrate species but has not been found in invertebrates. It is a member of the protocadherin family, the largest subgroup within the cadherin superfamily of large cell surface proteins characterised by a variable number of extracellular cadherin domains.16 The cadherins are involved in calcium-dependent cell–cell adhesion and are important in nervous system function.
Cadherin/protocadherin genes have previously been shown to be involved in disorders of the retina: mutations in the protocadherin gene PCDH15 are responsible for Usher syndrome type 1F, and mutations in the cadherin genes CDH23 and CDH3 were shown to cause Usher syndrome type 1D and hypotrichosis with juvenile macular dystrophy, respectively.17–19
PCDH21 has six predicted cadherin repeats and an intracellular domain, which is not homologous to that of other cadherins. PCDH21 is highly expressed the retina, more specifically in the junction between the inner and outer segments (OS) of rod and cone photoreceptors; the expression is also high in olfactory bulbs, with a lower expression in the septum and olfactory cortex.13 15 PCDH21 expression is very low or absent in heart, kidney, liver, lung, spleen, testis and skeletal muscle. The photoreceptor OS is a derivative of a non-motile cilium that contains phototransduction proteins organised in a stack of over 1000 discs. The discs are continuously renewed from the base, and the older discs are shedded from the tip. The proteins are synthesised in the inner segment and transported to the OS via the connecting cilium.
PCDH21 undergoes a specific proteolytic cleavage, which releases ectodomain as a soluble fragment. In rds knockout mice, where the OS assembly is disturbed, the proteolytic cleavage is inhibited and full-length PCDH21 accumulates.20
Mutations in PROM1 have been identified in patients with autosomal recessive RP, and PCDH21 protein has been shown to interact with PROM1 by co-immunoprecipitation studies.21 PROM1 is a transmembrane protein associated with plasma membrane protrusions, and it co-localises with PCDH21 to the base of the photoreceptor OS. In transgenic mice expressing human PROM1 with a heterozygous missense mutation, PROM1 and PCDH21 protein was found throughout the photoreceptor layers instead of the normal localisation at the base of the photoreceptor OS. In Pcdh21−/− mice, PROM1 mis-localised throughout the OS. These findings suggest that the interaction between PCDH21 and PROM1 is necessary for both proteins to function normally.
Taken together, these findings suggest that PCDH21 is involved in OS structure and/or assembly but not in phototransduction in rods and cones. This hypothesis is supported by the localisation of PCDH21 to the base of the OS at the earliest stages of differentiation of rods, before other OS proteins, for example, rhodopsin, RP1 and ROM1, are found.20
A PCDH21 knockout mouse model showed normal viability and fertility without anatomical or behavioural abnormalities.13 At age 1 month, the retinas appeared normal, but this was followed by a progressive loss of photoreceptors, and a 50% reduction in the number of photoreceptor cells was observed by age 6 months, which likely is the result of apoptosis. Analysis of photoreceptor electrical signalling showed 2- to 3-fold diminished saturating a-wave amplitude, which reflects rod photoreceptor function, in Pcdh21−/− mice compared to Pcdh21+/+ and Pcdh21+/− mice. The saturating amplitude of the cone-driven b-wave response was diminished about twofold in Pcdh21−/− mice. Electron microscopy showed that in Pcdh21−/− mice, the OS were shorter with swirls of membranes with imperfectly stacked discs. The inner segments and RPE appeared unaffected. The knockout mouse thus had abnormal cone and rod function, but the dysfunction of rods and cones was not as severe as expected from the degree of disorganisation of the OS. The ERG findings and the loss of photoreceptor cells in patients with PCDH21 mutations were similar to those found in the knockout mouse model. Despite the high PCDH21 expression in olfactory bulbs, the patients showed no signs of decreased olfactory function.
The frameshift mutation found in PCHD21, one of three sequenced genes of 11 candidate genes in the linkage interval, the segregation of the mutation with the disease in the family, the high and almost exclusive expression of the gene in the retina and the similarity of the phenotype of the patients with that of a knockout mouse heavily imply that autosomal recessive cone-rod dystrophy in this family is caused by PCDH21 mutations. Since several of the previously reported genes involved in CORD have been shown to be involved in, for example, Stargardt disease, Leber congenital amaurosis and RP, PCDH21 should be considered a candidate gene for other retinal dystrophies.
Note Around the time of the acceptance of this paper, PCDH21 mutations were reported in two families with retinal dystrophy in Henderson RH, Li Z, Abd El Aziz MM, Mackay DS, Eljinini MA, Zeidan M, Moore AT, Bhattacharya SS, Webster AR. Biallelic mutation of protocadherin-21 (PCDH21) causes retinal degeneration in humans. Mol Vis 2010;16:46–52.
Electronic database information
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for PCDH21 (accession number NM_033100)). Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for CORD).
Acknowledgments
We thank the family for their participation.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
Funding This work was supported by grants from The Danish Association of the Blind and The Danish Medical Research Council.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Faroese Ethical Committee.
Provenance and peer review Not commissioned; externally peer reviewed.