Article Text

Abstract
The inactivation of programmed cell death has profound effects not only on the development but also on the overall integrity of multicellular organisms. Beside developmental abnormalities, it may lead to tumorigenesis, autoimmunity, and other serious health problems. Deregulated apoptosis may also be the leading cause of cancer therapy chemoresistance. Caspase family of cysteinyl-proteases plays the key role in the initiation and execution of programmed cell death. This review gives an overview of the role of caspases, their natural modulators like IAPs, FLIPs, and Smac/Diablo in apoptosis and upon inactivation, and also in cancer development. Besides describing the basic mechanisms governing programmed cell death, a large part of this review is dedicated to previous studies that were focused on screening tumours for mutations within caspase genes as well as their regulators. The last part of this review discusses several emerging treatments that involve modulation of caspases and their regulators. Thus, we also highlight caspase cascade modulating experimental anticancer drugs like cFLIP-antagonist CDDO-Me; cIAP1 antagonists OSU-03012 and ME-BS; and XIAP small molecule antagonists 1396–11, 1396–12, 1396–28, triptolide, AEG35156, survivin/Hsp90 antagonist shephedrin, and some of the direct activators of procaspase-3.
Statistics from Altmetric.com
On average, one out of every four people will have cancer in their lifetime. Although inherited cancers account for only a small fraction of all tumours, most cancers are caused by a mix of hereditary and environmental factors.1 Identification of cancer stem cells in the majority of cancers suggests that the mutations are occurring within tissue stem cells, and cancer is both a consequence of uncontrolled proliferation, as well as disturbed differentiation.2 3 Genetic alterations often allow for verification of diagnosis and may even dictate the treatment approaches. Thus, cancer specific therapies based on specific genetic alterations (pharmacogenomics) have opened a new era of cancer treatment.4
Multicellular organisms employ two main mechanisms for the elimination of cells: necrosis and apoptosis.5 Necrosis may be triggered by the rupture of the plasmatic membrane and may be accompanied by formation of an inflammatory process.6 On the contrary, apoptosis involves a “cleaner” type of death, in which the chromatin is condensed; the DNA becomes fragmented forming vesicles known as “apoptotic bodies”. These are rapidly phagocytosed by the macrophages with the result that the cell disappears without any inflammatory phenomena.7 Apoptosis induction might be achieved in several ways—for example, by promoting the expression of pro-apoptotic factors while reducing the expression of anti-apoptotic factors only in the tumour cells, or by means of the infection of viral particles that act specifically within the transformed cells.8
In mammals, apoptosis can be initiated by three different pathways: (1) the extrinsic pathway, which can be triggered by ligation of death receptors and subsequent caspase-8 activation; (2) the intrinsic pathway, which is initiated by cellular stress followed by activation of caspase-9; or (3) the granzyme B pathway, where the cytotoxic cell protease granzyme B is delivered to sensitive target cells. Each of these pathways converges to a common execution phase of apoptosis that requires proteolytic activation of caspases-3 and/or -7 from their inactive zymogens.9 10 Biochemically, the main features of apoptosis include caspase cascade activation and DNA fragmentation.11 Mitochondria also play a key role in mediating apoptosis induced by diverse stimuli. They release pro-apoptotic proteins (cytochrome c, Smac, Omi, AIF, and EndoG) whose release into the cytosol is regulated by proteins belonging to the Bcl2 family.
EXTRINSIC AND INTRINSIC APOPTOSIS PATHWAY
The receptor triggered or extrinsic apoptotic pathway was the first one to be described (fig 1). The receptors triggering this pathway are located in the cell membrane and they are activated by extracellular ligands. Typical death receptors are Fas (fibroblast associated antigen, also called Apo-1 or CD95) and tumour necrosis factor receptor (TNF-R) 1; they belong to TNF-R family and contain a cytosolic death domain (DD). Ligation of death receptor causes formation of death inducing signalling complex (DISC)12 13 in which the adaptor proteins FADD and/or TRADD bind with their death domain (DD) to a DD in the cytoplasmic region of the receptors.14 The receptor induced pathway leads to the recruitment of caspase-8 or -10 (initiator caspases) to the DISC.15 The activated caspase then proteolytically activates downstream effector caspases (also called executioner caspases) that degrade cellular targets. In accordance with a pivotal role of FADD and caspase-8 in CD95- or TRAIL induced cell death, mice or cell lines deficient in these molecules are completely protected from the apoptotic action of TRAIL or CD95L.16–18 Activated caspase-8 then directly cleaves pro-caspase-3 or other executioner caspases, eventually leading to the apoptosis. Caspase-8 can also cleave the BH3-only protein Bid. The resulting truncated Bid (tBid) then moves to the mitochondria and induces cytochrome c release, leading to activation of caspase-9 and caspase-3. DISC signalling can be inhibited by expression of c-FLIP, a physiologic dominant negative caspase-8 that leads to the formation of a signalling inactive DISC.19
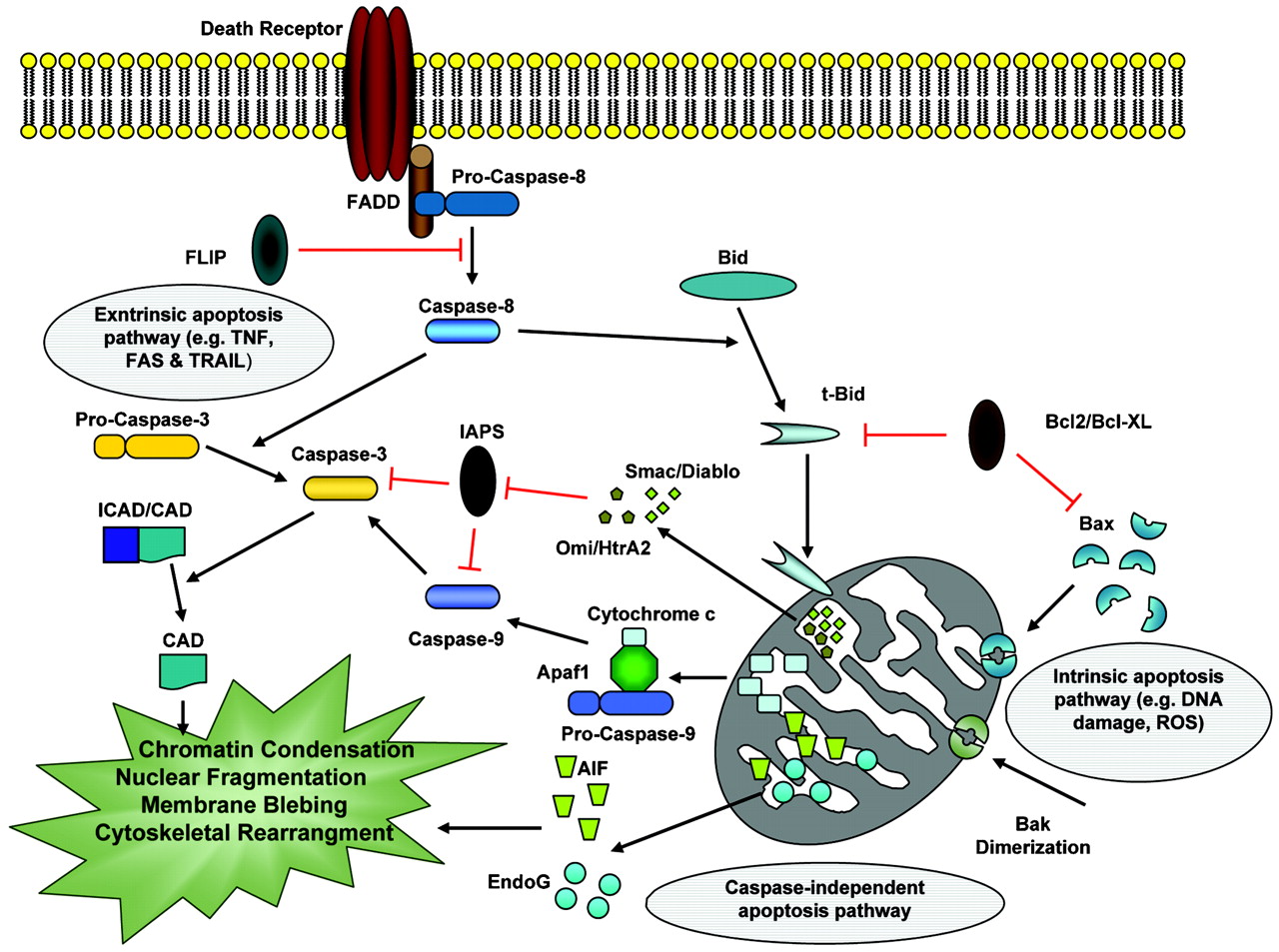
{kind=link}
The molecular mechanisms of apoptosis. Apoptosis pathways can be initiated via different stimuli—that is, at the plasma membrane by death receptor ligation (extrinsic pathway) or at the mitochondria (intrinsic pathway). Stimulation of death receptors results in receptor aggregation and recruitment of the adaptor molecule Fas-associated protein with death domain (FADD) and caspase-8. Upon recruitment, caspase-8 becomes activated and initiates apoptosis by direct cleavage of downstream effector caspases. Mitochondria are engaged via the intrinsic pathway, which can be initiated by a variety of stress stimuli, including ultraviolet (UV) radiation, γ-irradiation, heat, DNA damage, the actions of some oncoproteins and tumour suppressor genes (that is, p53), viral virulence factors, and most chemotherapeutic agents. Mitochondrial membrane permeabilisation is regulated by balance of opposing actions of proapoptotic and antiapoptotic Bcl2 family members (Bax, Bak, Bcl2 and Bcl-XL, Mcl-1). Following mitochondrial permeabilisation, mitochondrial pro-apoptotic proteins like cytochrome c, Smac/Diablo, Omi/HtrA2 (caspase dependent), AIF, and Endo G (non-caspase-dependent) release via transmembrane channels across the mitochondrial outer membrane (see main text for more details). CAD, caspase activated DNase; FAS, fibroblast associated antigen; ICAD, inhibitor of CAD; ROS, reactive oxygen species; TNF, tumour necrosis factor; TRAIL, TNF related apoptosis inducing ligand.
The intrinsic or mitochondrial pathway is activated by a variety of extra- and intracellular stresses, including oxidative stress, irradiation, and treatment with cytotoxic drugs (fig 1).20–22 Unlike the death receptor dependent pathway, the mitochondria dependent pathway is mediated by Bax/Bak insertion into mitochondrial membrane, and subsequent release of cytochrome c from the mitochondrial inter-membrane space into the cytosol.23 Anti-apoptotic Bcl-2 family members, such as Bcl-2 and Bcl-XL, prevent cytochrome c release, presumably by binding and inhibition of Bax and Bak. BH3-only proteins, such as Bid and Bim, contribute to the pro-apoptotic function of Bax or Bak by inducing homo-oligomerisation of these proteins. Cytochrome c then binds to the Apaf1 and together with (d)ATP causes recruitment of pro-caspase-9 to the complex.24–26 The formed multi-protein complex is called apoptosome, which contains several units of Apaf1 and other above molecules and, depending on the isolation method, it has between 700 kBa –1.4 mBa.27–31 Activated caspase-9 in turn activates caspase-3 and initiates the proteolytic cascade.32 In addition to cytochrome c, mitochondria release a large number of other polypeptides, including AIF,33 Endo G, second mitochondrial activator of caspases (Smac/DIABLO)34 and HtrA2/Omi35 from the intermembrane space. Smac/Diablo and Omi/HtrA2 promote caspase activation through neutralising the inhibitory effects of inhibitor of apoptosis proteins (IAPs),36 while AIF and endonuclease G cause DNA damage and condensation.37
CASPASE FAMILY
The first identified caspase, caspase-1, a homologue of CED-3, was actually interleukin-1β processing enzyme (ICE), originally discovered in totally different biologic context38 39 (see below). Overexpression of ICE may induce or sensitize to apoptosis,38 therefore it was suggested that mammalian caspases may have an essential apoptotic functions as CED-3 in Caenorhabditis elegans (C elegans) cells. The official nomenclature names 14 caspases in mammals.19 Caspase-1, -10, and -14 were found in human.40 41 Caspase-13 was later proved to be a bovine homologue of human caspase-4.42 Caspase-11 and -12 are murine homologue enzymes of human caspase-4 and -5.43 Not all the caspases are involved in programmed cell death, and not all forms of cell death require caspases. Indeed, some caspases are crucial for apoptosis, some are not necessary, and most caspases have functions other than just take part in executing apoptosis, like cell survival, proliferation, differentiation or inflammation.44–46
The function of caspase is very closely related to its structure. Different caspases show different substrate preferences, although aspartate at P1 position is universally required for all caspase substrates. Some caspases have long prodomains containing special motif such as DED (caspase-8 and -10), and caspase recruitment domains (CARD) (caspase-1, -2, -4, -5, -9, -11 and -12), which allow for interactions with other proteins, and link with signalling pathways.
Mechanisms of caspase activation
Caspases are synthesised as a single chain of inactive zymogens, consisting of four domains: an N-terminal prodomain of variable length, a large subunit with a molecular weight of about 20 kDa, a small subunit (∼10 kDa), and a linker region connecting these catalytic subunits.47 The linker region is missing in some family members. Proteolytic cleavage of the caspase precursors results in the separation of large and small subunits with the production of a hetero-tetrameric complex (the active enzyme) consisting of two large and two small subunits.48 Caspases differ in the length and in the amino acid sequence of their N-terminal prodomain. The long prodomain (more than 90 amino acid residues) contains one of two modular regions essential for the interaction with adaptor proteins. These modules contain DED or CARD. Hydrophobic protein interactions are mainly achieved via DED-DED contacts, whereas electrostatic interactions occur through CARD-CARD contacts.47 Based on their proapoptotic functions, the caspases have been divided into two groups: initiators and effectors. First group of initiator (or apical) caspases (caspases-2, -8, -9, -10, and, probably, -11) activate the second-group of caspases (caspases-3, -6, and -7). The effector (or downstream) caspases are able to directly degrade multiple substrates including the structural and regulatory proteins in the cell nucleus, cytoplasm, and cytoskeleton.49 In some cases, initiator caspases can also function as effector caspases; this activity helps to amplify a suicide signal in the cell whose death pathways have been only weakly initiated. Furthermore, the activation of effector caspases can not only be caused by initiator caspases, but also by other, non-caspase proteases, including cathepsins, calpains, and granzymes. Caspase-1 and caspase-4, -5 have similar structures and are predominantly involved in the maturation of proinflammatory cytokines. However, significant bodies of experimental evidence exist that indicate a redundant/accessory role of these caspases in apoptosis.38 50 The caspase proteolytic signalling cascades are interconnected and due to overlapping substrate specificity they are also partially redundant. As a result, the apoptotic signal can be significantly amplified. A number of cellular and viral caspase inhibitors exist that may prevent both initiation and amplification of the apoptotic signal within the proteolytic cascade.47 Below, we briefly introduce selected caspases.
Caspase-2
Caspase-2 is the second identified caspase. It contains a CARD-domain and recruit multi-protein complex “PIDDosome2 through CARD binding with RAIDD (RIP associated ICH-1/ECD3 homologous protein with a death domain).51 The adaptor proteins RAIDD and PIDD (p53 induced protein with death domain) in this complex are essential components for the activation for caspase-2.52 The function of capase-2 is still poorly understood. While containing the long prodomain and being able to respond to a variety of apoptotic stimuli,53 and RAIDD mediated interaction with Fas,43 54 it also bears some characters unlike an initiator. The substrate preference of it is more close to caspase-3 and -7, and it can even be activated by caspase-3, a downstream executioner. One more unique feature of caspase-2 is the localisation to the nucleus and the Golgi apparatus in addition to cytosol.55–57 However, caspase-2 appears to act upstream of mitochondrial permeabilisation by cleaving and activating Bid, and plays an important role for DNA damage induced apoptosis.58–60 In response to genotoxic stress, the activation of either caspase-2 or NF-κB is controlled by different isoforms of PIDD, and will respectively lead cell to apoptosis or survival.61 However, in caspase-2 KO animal models, caspsase-2 is not really essential for most physiological cell deaths. A recent study demonstrated that disruption of caspase-2 has a significant impact on mouse aging, suggesting that caspase-2 deficiency compromised the animal’s ability to clear oxidative damaged cells.62 Caspase-2 may have more distinctive properties, and understanding the function of caspase-2 is challenging. In a T-2 toxin induced apoptosis model, caspase-2 activation is observed earlier than all the other caspases, and caspase-2 might even affect caspase-8 activation.63
Caspase-8
The function of caspase-8 is well established. It is essential for the extrinsic cell death pathways initiated by the TNF family members.64 Death receptors will recruit the DISC upon binding specific TNF family ligands and trimerisation. Procaspase-8 can be recruited into this complex via the adaptor protein FADD. The dimerised, or trimerised, procaspase-8 molecules in the DISC are activated through reciprocal cleavage. Activated caspase-8 then initiates downstream apoptotic cascade by cleaving caspase-3, caspase-7 or Bid.65–67 The activated caspase-8 can also activate NF-κB and regulate lymphocyte proliferation. In proliferating cells, caspase-8 remains largely unprocessed, and becomes only weakly activated, while in FasL induced apoptosis, caspase-8 processing and strong activation is observed. The key protein for regulating caspase-8 activation level is c-FLIPL, a caspase-8-like molecule that lacks caspase activity.68 69 In the absence of c-FLIPL, the dimerisation or trimerisation of procaspase-8 leads to full processing and activation of procaspase-8 molecules. At low concentrations of c-FLIPL, procaspase-8 preferably forms heterodimers with c-FLIPL. Then limited procaspase-8 activation occurs and active heterodimers remain associated with the DISC complex. Depending on the specific subset of substrates that is cleaved, either apoptosis or NF-κB activation can ensue. At high concentrations of c-FLIPL, procaspase-8 recruitment is blocked, and c-FLIPL cleavage is ensured by basal caspase-8 activity, and subsequently NF-κB is activated.70 71 In fact, besides function in TNF family induced apoptosis pathway, caspase-8 also have other non-apoptotic functions such as in the macrophage differentiation, T cell, B cell and NK (natural killer) cell proliferation, and heart muscle development.40 44 45
Caspase-9
Caspase-9, the apical/initiator caspase within the apoptosome dependent cascade, has been extensively studied within the last 12 years. When the mitochondrial pathway is activated, cytochrome c is released from the mitochondria, and is recruited to the cytoplasmic receptor, Apaf1.72 73 In the presence of dATP or ATP, cytochrome c and Apaf1 assemble into a complex called “apoptosome”. Procaspase-9 then binds to Apaf1 through their CARD domain and becomes activated by reciprocal interaction with another procaspase-9.74 75 Then activated apoptosome bound caspase-9 cleaves and activates downstream enzyme, caspase-3.
Caspase-10
Like caspase-8, caspase-10 possesses two DEDs domain, which can be recruited to the same DISC as caspase-8 and activated by death receptors. It can also cleave Bid and activate the mitochondrial pathway, suggesting that it may have an overlapping function with caspase-8. Despite the fact that it is highly homologous to caspase-8 both in structure and function, there is no mouse homologue of caspase-10.43 76 77 The association between caspase-10 gene dysfunction and an autoimmune disease ALPS-2, suggests that caspase-10 might have some different roles from caspase-8 in some cell types. However, the contributions of mutation to the disease are not conclusive.76 78
Caspase-12
Caspase-12 is a murine caspase containing a CARD prodomain. Its human counterpart, also caspase-12, appears to be non-functional due to gene mutation.79 Unlike other caspases, caspase-12 is localised specifically to the endoplasmic reticulum (ER), and is a specific sensor responding to ER stress induced cell death.80 81 In some cell types, under ER stress (usually caused by the accumulation of proteins), translocation of caspase-7 from the cytosol to the ER surface had been observed. Procaspase-12 is then activated as an initiator caspase, which can lead to procaspase-9 activation. In this kind of caspase-9 mediated pathway, cytochrome c is not involved in the activation of procaspase-9.82–84 Murine caspase-12 shows homology to human caspase-4, which also had been proved to be involved in ER stress induced apoptosis,85 but it also plays a role in the immune system (see below).
Caspase-1, -4, -5, -11
Caspase-1, -4, -5, -11 formed the so-called “inflammatory caspases” group. Caspase-11 is a murine enzyme sharing lots of similarity with both caspase-4 and -5. It might be an ancestor gene to the other two caspase genes.46 These caspases are termed “inflammatory” as the main caspase-1 substrates identified to date are proIL-1β and proIL-18, two related cytokines that play critical roles in inflammation.46 86 Targeted deletion of caspase-1 had no effect on animal development, and the embryonic fibroblasts and thymocytes from these mice are still very sensitive to various apoptotic stimuli.87 Caspase-1−/− mice had, however, major defects in the production of mature IL-1β and impaired IL-1α synthesis. Secretion of TNF and IL-6 in response to LPS (lipopolysaccharide) stimulation was also reduced. In addition, macrophages from caspase-1−/− mice were defective in LPS induced IFN-γ production.88 Caspase-1−/− mice were also resistant to the lethal effects of LPS.89 Thus, based on these murine models, the apoptotic function of caspase-1 seems not to be as necessary as it is in inflammatory reaction. Since caspase-4 and -5 are non-existent in mice, no direct data on their targeted disruption could be obtained. However, other caspases could also be involved in immunoregulation. It has been demonstrated that caspase-11 can process caspase-3 directly during ischaemia and septic shock in addition to regulating caspase-1 activation.90
Caspase-3, -6, -7
These three effector caspases are highly homologous to each other.43 Their final functions are also similar in executing apoptosis. While caspase-3 has been extensively studied, we have much less knowledge about caspase-6 and -7. None of them can fully control the execution of all the aspects of apoptosis. The contribution of each caspase to the cell death or dysfunction could be varied as well. In apoptotic cells, caspase-3 is the main executioner as it can be activated through both extrinsic and intrinsic signalling pathway, but it cannot be cleaved by caspase-2.91 Moreover, depletion of caspase-3 in cell-free apoptotic system cause inhibition of various downstream events while depletion of either caspase-6 or caspase-7 do not show any effect.92 Caspase-3 might be more important in most of downstream affairs, yet caspase-6 and –7 may have distinct roles in specific pathways, such as the special function of caspase-7 in ER stress induced apoptosis.84
MUTATIONS WITHIN THE CASPASE FAMILY AND CANCER
Malfunction of apoptosis plays an important role in the pathogenesis of tumours. Tumour cell survival could be induced by inactivation of proapoptotic signalling or activation of antiapoptotic pathways. There are two major ways that could downregulate cancer cell apoptosis: (1) somatic and non-somatic mutation and loss of expression of proapoptotic molecules; and (2) overexpression of apoptosis inhibitory molecules.93 94 Somatic mutations of apoptosis related genes affect several proteins. Mutations within caspase family proteases are not uncommon in malignancies.95 Here we focus on some frequent mutations within caspase family and their proposed role in different cancers.
Mutations of caspase-8 gene and cancer
Several reports show that caspase-8 is mutated in different types of cancers. Soung and colleagues94 screened gastric carcinomas (162 cases), breast carcinomas (93 caspase), non–small cell lung cancers (NSCLC) (185 cases), and 88 acute leukaemias (88 cases) for mutations within the caspase-8 gene using single strand conformation polymorphism (SSCP).94 Interestingly, the caspase-8 mutations were mostly detected in gastric cancers but not in other cancer types. They found that the incidence of caspase-8 mutation in gastric cancer is statistically higher than those of NSCLC, breast cancer, and acute leukaemias. Furthermore, all of the 13 mutations detected were in advanced gastric cancers but not in early gastric cancers. They reported that in 122 analysed advanced gastric cancers, 13 (10.1%) cancer samples harboured caspase-8 mutations. The mutations consisted of three missense, one in-frame deletion, and five frameshift mutations in the coding sequences; two mutations in the initiation codon; three mutations in the introns; and one mutation in the 3V untranslated region (table 1). The missense mutations detected in this study would result in the substitution of amino acids in the DED and the p10 subunit. The frameshift mutations would result in premature terminations of caspase-8 protein synthesis (table 1). They also proved that the caspase-8 mutants were expressed well and the sizes of the detected mutants were matched with the predicted amino acid changes using in vitro translation and subsequent immunoblotting. They transfected 293T, 293, and HT1080 cells with observed caspase-8 mutants. All mutants showed significant decrease of caspase-8 activity in apoptosis induction compared with the wild type caspase-8 (except mutation 1427T >C).94
In another study, the sensitivity of NSCLC and small cell lung carcinoma (SCLC) cells to the death receptor dependent cell death was investigated using cell lines derived from patients (they used the same concentrations of FasL and TRAIL in all experiments).96 While most NSCLC cells expressed detectable amounts of surface Fas, TRAIL-R1 and R2, they could not detect any surface death receptors in tested SCLC cell lines. They also reported that caspase-8 protein precursor was undetectable in SCLC cells, but they do not elaborate on the nature of the defects in its expression.96
In another study, in breast cancer, D302H substitution was reported as a widespread variant in the caspase-8 gene.97 However, a large multi-ethnic cohort study (MEC) that included over 215 000 men and women in Hawaii and California, comprising predominantly self declared African Americans, Japanese Americans, native Hawaiians, Latinos and European Americans who were between the ages of 45–75 years at enrolment,98 99 found no significant inverse association between D302H variant in the caspase-8 gene and risk of three common cancers (breast, colorectal, prostate) in pooled analyses.98 They included groups of various ancestral backgrounds with very different disease risks and allele frequencies and consisted of large size and multiple cancer end points. Thus, the likelihood that the lack of significant association was attributable to bias or population stratification was unlikely.98
In another study, caspase-8 was investigated in meningioma.100 Their study was based on five case–control series that contributed to the international Interphone study.101 Briefly, the Interphone Study was a multicentre epidemiological case–control study to investigate whether mobile phone usage increases the risk of primary brain tumour (PBT) and malignant parotid gland tumours. The five case–control series of PBTs were assembled in the Thames regions of Southeast England; the Northern UK including central Scotland, the West Midlands, West Yorkshire, and the Trent area; the Stockholm, Lund, Göteborg, and Umeå regions of Sweden; throughout Denmark; and in all regions of Finland except Northern Lapland and Åland.101 They could not find any significant increase in risk of meningioma and caspase-8 D302H. Their results were not consistent with other previously published data on meningioma that showed increased risk of caspase-8 D302H variant on meningioma development.102 Again, this controversy might be due to population and ethnicity of the corresponding studies.
A case–control study in a Chinese population was done to evaluate the associations of caspase-8 mutation and pancreatic cancer.103 This study consisted of 397 patients with pancreatic cancer and 907 controls. All subjects were Han Chinese. Genotypes of caspase-8 -652 6N in/del polymorphisms were determined in this study. Caspase-8 -652 6N del/del genotypes showed a multiplicative joint effect with FasL and Fas in attenuating susceptibility to pancreatic cancer. The caspase-8 -652 6N in/del polymorphisms are summarised in table 2. Glioma accounts for about 80% of malignant primary brain tumours.104 Hypermethylation of caspase-8 has been linked with glioblastoma multiforme relapse,105 suggesting that caspase-8 may have a role in the development of glioma. It was found that D302H was also a risk determinant of glioma.106
On the other hand, Sun et al107 identified a 6 bp deletion polymorphism (−652 6N del) in the promoter of the CASP8 gene that abolishes the binding of Sp1 transcription factor and was associated with the decreased RNA expression in lymphocytes, and lower caspase-8 protein level. This deletion variant was found to be associated with an approximately 25% increased risk (per copy) of lung, oesophageal, stomach, colorectal, breast and cervical cancer in a Chinese population (4938 cases and 4919 controls).107 This is, however, contradicted by analysis of four breast cancer case–control studies where data on various mutations including caspase-8 mutation were analysed.108 These studies included: German Familial Breast Cancer Study (GFBCS), index patients of 1110 German BC families and 1108 control individuals, Sheffield Breast Cancer Study which included white Anglo-Saxon Sheffield residents, including 1212 pathologically confirmed patients with prevalent and invasive breast cancer recruited and 1184 unselected cancer-free women attending the Mammography Screening Service,109 110 Gene Environment Interaction and Breast Cancer in Germany (GENICA) in which 1143 incident breast cancer cases and 1155 population controls were recruited from the Greater Bonn Region, Germany,111 112 and finally studies of Epidemiology and Risk Factors in Cancer Heredity (SEARCH) which included breast cancer and control subjects.113 They could not find any association between caspase-8 -652 6N del promoter polymorphism and breast cancer. Given this lack of association in Europeans, it was suggested that the functional caspase-8 6N del promoter variant may have an ethnicity specific effect due to different genetic backgrounds (Asians vs Europeans) and it could interpret the findings that Sun et al reported of the association of this polymorphism with breast cancer.107
Pancreatic cancer is one of the leading causes of cancer related death in the world.114 115 Smoking, diabetes mellitus history, and, perhaps, alcohol drinking are risk factors for pancreatic carcinogenesis.116–119 However, only a part of exposed individuals develops pancreatic cancer in their lifespan, suggesting that genetic susceptibility factors also play a role in pancreatic carcinogenesis. It has been shown that pancreatic cancer cells often present non-functional CD95/Fas and aberrant expression of FasL, and this mechanism may contribute to the malignant and often rapid course of the disease.120–122 It is unclear if CD95/Fas itself or downstream signalling molecules like caspase-8 are inactivated in these cases.
Caspase-9 gene mutation and cancer
Caspase-9 is a virtually ubiquitous protease, constitutively expressed in a variety of fetal and adult human tissues.123 124 Mutational analysis of caspase-9 was performed in neuroblastoma tissues; however, no somatic mutation of caspase-9 in the tumours were found.125 In another study 180 gastric, 104 colorectal and 69 lung adenocarcinomas were randomly selected for the study.126 They isolated genomic DNAs from normal and tumour tissues of the same patients and studied the entire coding region with all splice sites of the caspase-9. Silent mutations were detected in two colorectal carcinomas and one gastric carcinoma. The mutations consisted of a G-to-A transition at nucleotide 261 (261G>A; S87S) in exon 2, a G-to-A transition at nucleotide 588 (588G>A; S196S) in exon 4, and a G-to-A transition at nucleotide 1101 (1101G>A; L367L) in exon 8.
In a case–control study, lung cancer patients and age and gender matched healthy controls were investigated for caspase-9 promoter polymorphism in lung cancer.127 All cases and controls were ethnic Koreans in this study and the cases included 210 (48.6%) squamous cell carcinomas, 141 (32.6%) adenocarcinomas, 73 (16.9%) small cell carcinomas, and eight (1.9%) large cell carcinomas.127 They observed a significant difference in the distribution of the -2712C>T genotypes between the cases and controls, but there was no significant difference in the distribution of genotypes between cases and controls for -21263A>G, -2905T>G, and -2293del.127 They reported that the -21263 GG genotype was associated with a significantly decreased risk of lung cancer compared with the -21263 AA or the combined 21263 AA + AG genotype. They also found that for the 2712C>T polymorphism, individuals with at least one -712T allele were at a significantly increased risk of lung cancer compared with those harbouring -712 CC genotype, and the risk of lung cancer increased with increasing numbers of -712T alleles. Their other finding showed that the -905T>G and -293del polymorphisms were not significantly associated with the risk of lung cancer.127 Another interesting finding of this study was that caspase-9 polymorphisms and their haplotypes interacted with tobacco smoking. They found that caspase-9 polymorphisms were significantly associated with the risk of lung cancer in the smokers but not in the non-smokers, which reflects a gene–environment interaction. Such an interaction is biologically plausible because smoking is a major risk factor for lung cancer. It was also found that the association between caspase-9 polymorphisms and the risk of lung cancer was statistically significant in the light smokers but not in the heavy smokers. Thus, the data indicate that an environmental carcinogen (components of cigarette smoke), if applied in larger quantities, may override genetic predisposition.128 129 In another study, caspase-9 polymorphism was investigated in multiple myeloma in a case–control study (183 patients and 691 controls).130 Genotyping of the caspase-9 single nucleotide polymorphism (SNP) [Ex5þ32 G>A (rs1052576)] was done in this study. They found a protective association for the mentioned caspase-9 polymorphism in multiple myeloma.
Mutations within caspase-3 and cancer risk
Caspase-3 is an effector caspase, and is activated by extrinsic and intrinsic cell death pathways. It plays a central role in the execution phase of cell apoptosis.28 76 131 A caspase-3 mutation has been reported in the MCF-7 breast cancer cell line,132 suggesting the presence of caspase-3 mutation in human cancer tissues. There are several recent reports that have focused on the caspase-3 mutation in different cancers. Soung et al have investigated caspase-3 somatic mutation in several cancers.133 They investigated 165 stomach carcinomas, 95 colon carcinomas, 76 breast carcinomas, 80 hepatocellular carcinomas, 181 non-small cell lung cancers, 28 multiple myelomas, 12 medulloblastomas, 15 Wilms’ tumours, 12 renal cell carcinomas, 40 oesophagus carcinomas, 33 urinary bladder carcinomas, 33 laryngeal carcinomas, 129 non-Hodgkin lymphomas, and 45 acute leukaemias and compared healthy and malignant tissue from the same patients for caspase-3 somatic mutations. They did not observe any evidence of mutations in normal samples from the same patients and concluded that the mutations had risen somatically.133 The mutation data are summarised in table 3, and show that caspase-3 was mutated in one case in stomach adenocarcinoma, one case in lung cancer, four cases in colon cancer, one case in hepatocellular carcinoma, and one case in multiple myeloma. The mutations consisted of six missense mutations, four silent mutations, two mutations in the introns, one mutation in the 5′-untranslated region, and one mutation in the 3′-untranslated region. Of the six missense mutations, two were predicted to involve the p17 large protease subunit and the other four to involve the p12 small protease subunit. Of note, two missense mutations in exon 6 showed an identical A to T transversion at base pair 674 in unrelated individuals.
In other study, caspase-3 mutation was investigated in squamous cell carcinoma of the head and neck (SCCHN). This case–control analysis included 930 patients with histologically confirmed SCCHN. All cases were non-Hispanic whites and had not received any radiotherapy or chemotherapy at the time of recruitment and blood donation. The 993 cancer-free subjects were recruited in the same time period, who were frequency matched to the cases by age, sex, and ethnicity.134 SCCHN is one of the most common cancers in the world.135 It is estimated that there were approximately 40 566 new cases of SCCHN in the USA in 2007.114 They identified caspase-3 transcriptional regulatory region (rs4647601:G>T, rs4647602:C>A, and rs4647603:G>A) polymorphisms in case and control subjects.134 The distribution of these polymorphisms in case and control group is shown in table 4. There was no statistically significant difference in the distributions of either allele or genotype frequencies of these three SNPs. However, when comparing with the GG genotypes of the caspase-3 rs4647601:G>T, they found an association of the caspase-3 rs4647601:TT genotype with an increased risk of SCCHN, and there was no association with any genotype of the caspase-3 rs4647602:C>A and rs4647603:G>A SNPs (p>0.180, p>0.547). They also found evidence that caspase-3 rs4647601:TT genotype increased the risk of SCCHN in the subgroups of younger males, non-drinkers and never-smokers.134
Caspase-7 mutations and oncogenesis
Caspase-7 is another effector caspase that is comparatively important to caspase-3 in apoptosis execution, especially in the cells with deficient or under-expressed caspase-3.136 137 As other protease family members, caspase-7 is expressed as an inactive proenzyme, that upon processing generates a large p23 and a small p12 subunit.40 138 Caspase-7 somatic polymorphisms were studied in some common cancers. Briefly, 33 SCCHN, 35 transitional cell carcinomas of urinary bladder, 50 oesophageal squamous cell carcinomas, 80 non-small cell lung cancers, 98 colon adenocarcinomas and five gastric adenocarcinomas were compared with corresponding normal cells from the same patients.139 The somatic polymorphism of caspase-7 in these patients is illustrated in table 5. Among the mutations of caspase-7 found in this study, one nonsense mutation (colon cancer, exon 2, 127 C to T) was identified in the coding regions of the large p23 subunit. The nonsense mutation was predicted to cause premature termination of protein synthesis, and hence resemble typical loss-of-function mutations. The C to T transition at bp 127 leads to a termination at codon 43, resulting in a protein that has a prodomain and a part of p23 large subunit. In addition, the Arg at amino acid 43 is conserved in caspase-7 among the species of human, mouse and rat.139 This amino acid is also conserved in other caspases such as caspase-3, -6, -8 and -9.139 The Arg-43 of caspase-7 is one of the constituting residues for substrate’s P1 binding pocket,140 and it seems that alteration of the Arg-43 decreases the protease function of caspase-7. It was found that this mutant has a defect in induction of apoptosis.139
Mutations of inflammatory caspases -1, 4, and -5 and cancer
Pro-interleukin–1β and pro–interleukin-18β are the most important caspase-1 substrates which play critical roles in inflammation. There are several reports that inflammation plays important roles in cancer.141 142 Cancer cells produce many inflammatory mediators and interconnect with surrounding cells.142 Soung et al has recently investigated the probable role of inflammatory caspases in cancer.142 They identified somatic mutation for caspase-1, 4, and -5 in 337 patients with different cancers (103 colon carcinomas, 60 breast ductal carcinomas, 60 hepatocellular carcinomas, 54 gastric carcinomas, and 60 non-small cell lung cancers). They detected caspase-1 mutations in two (0.6%) of the 343 cancers, both in gastric carcinomas (54 samples) (3.7%) (table 6). The mutations consisted of one missense mutation in exon 7 (1034T>A) and one substitution mutation in intron 2 (IVS2−3C>A). They could not find any significant correlation of the caspase-1 mutations with histologic subtype of the gastric carcinomas.142 Caspase-4 mutations were detected in two (0.6%) of the 337 cancers, both in colon carcinomas (103 samples screened) (1.9%) (table 6). They could not identify any significant association between tumour size, metastasis, location of the tumours, patients’ sex/age, and recurrence and caspase-4 mutations.142 Caspase-5 mutations were detected in 15 (4.4%) of the 337 cancers. According to the tumour types, mutations were detected in nine gastric carcinomas (16.7%), four colon carcinomas (3.9%), one invasive ductal breast carcinoma (1.7%), and one lung adenocarcinoma (1.7%). The caspase-5 mutations consisted of nine mutations in exons, five mutations in introns, and one mutation in 5′-untranslated region (UTR). The nine mutations in the exons consisted of seven frameshift mutations in the (A)10 repeat sequences in exon 2, one missense mutation, and one silent mutation. According to tumour subtypes in the gastric carcinomas, eight caspase-5 mutations were detected in advanced gastric carcinomas (18.2%), while one mutation was observed in early gastric carcinoma (10.0%). There was no significant association of the caspase-5 mutation incidence with age/sex, stage, and histologic type of tumour.142
CASPASES AND CASPASE INHIBITORS: POTENTIAL CLINICAL APPLICATIONS
Since caspases play a central role in apoptosis and inflammation, they are attractive targets for diseases associated with uncontrolled cell proliferation as in cancer or autoimmune diseases. Thus, the controlled activation or inhibition of caspases offers an attractive means of therapeutic intervention. Below, we provide examples of such interventions, at various stages of implementation.
As indicated above, mutations within caspase family proteases are not uncommon in malignancies.95 Modulation of caspase activity for therapeutic purposes has been approached experimentally. Restoration of procaspase-3 expression both in respective cell lines as well as in primary tumour cells increases their sensitivity towards anticancer therapies. On the other hand, inhibition of the expression of some caspases—that is, by antisense RNA strategies—makes such cells more resistant towards classical chemotherapy.143 Efforts are being made to gain better understanding of the mechanisms responsible for activation of the caspase cascade so that anticancer drugs could be designed that directly target caspase activation mechanism rather than causing cellular stress, which would then lead to cell death.144 Still, caspases may not be the critical determinants of tumour’s sensitivity to cancer therapy (see below). Several studies show that tumour cells with a normal caspase activity are more responsive to anticancer-treatment,145 while some data indicate no correlation.146
Caspase inhibitors have been tested in other conditions besides cancer. For example, the development of intraepidermal blisters is a symptom of the autoimmune skin disease Pemphigus foliaceus caused by acantholysis and pathogenic autoantibodies against desmoglein 1, and could be prevented by application of Ac-DEVD-cmk, a peptide based caspase-3/7 inhibitor, and Bok-D-fmk, a broad spectrum caspase inhibitor.147 A Chinese traditional medicine (Shenfu injection), which consists mostly of ginsenocides and aconitine, suppresses apoptosis during hypoxia/reoxygenation in cardiomyocytes by increasing Bcl-2 expression and decreasing caspase-3 activity.148 Similarly, the treatment of tubular cell deletion in renal scarring with the pan-caspase inhibitor Bok-D-fmk markedly prevented renal proximal tubular cell apoptosis induced by cisplatin (rat model).149
c-FLIP and caspase-8
In some malignancies as previously mentioned, the gene encoding for caspase-8 is mutated or deleted or the expression of caspase-8 is altered so that it cannot bind to FADD conferring resistance to TRAIL (TNF-related apoptosis-inducing ligand) mediated cell death.150 TRAIL is a member of TNF-superfamily and belongs to the type II trans-membrane family of proteins; however, physiologically, like TNF, it is mostly active in its soluble trimeric form.151 It induces apoptosis by binding to death receptors present on the target cells, namely TRAIL-R1/DR-4 and TRAIL-R2/DR-5.152 An interesting feature of TRAIL is that it induces apoptosis predominantly in cancer cells while sparing normal cells.153 For example, primitive neuro-ectodermal brain tumour cells are resistant to TRAIL mediated apoptosis due to the loss of expression of caspase-8.154 Another example is human neuroblastoma malignancies that lack caspases-3 and -8, thereby resulting in resistance to standard chemotherapies.155 In some cases resistance to apoptotic cell death is associated with inactivation of caspase-8 gene by methylation.156
Apart from the regulation of caspase-8 expression, physiologic caspase-8 inhibitors also exist. Cellular “FLICE-like inhibitory protein” (cFLIP) is a cytosolic FLICE/casapase-8 inhibitor that, depending on splice variant may exist in its long form (cFLIPL), and short (cFLIPs) form. Because of cFLIP’s very high sequence homology to caspase-8, it competes with caspase-8 for binding to FADD within DISC and thereby prevents the binding of caspase-8 to FADD. This interaction of cFLIP with FADD prevents the receptor mediated apoptosis. Elevated expression of cFLIP has been observed in several kinds of tumours. For example, cFLIP is overexpressed in prostrate cancer, cervical cancer, ovarian cancer, colorectal cancer, gastric cancer, pancreatic cancer, and B cell chronic lymphocytic leukaemia.157 There are many reports indicating that downregulation of cFLIP sensitises tumour cells to apoptosis. For example, a recent finding has shown that cFLIP is a key regulator in colorectal cancer. siRNA mediated inhibition of cFLIP, induced apoptosis in p53 wild type, mutant and null colorectal cancer cells.158 Apart from that, intra-tumour delivery of siRNA duplexes induced apoptosis in xenografts of SCID mice. These experimental findings provide evidence that targeting of cFLIP in colorectal cancers can provide a potential therapeutic target.
Beside antisense oligonucleotides that target cFLIP, several small molecule inhibitors have the potential to downregulate the expression of cFLIPs, like DNA targeting anticancer drugs cisplatin and doxorubicin.157 Additionally, there are RNA synthesis and histone deacetylase inhibitors which can potentially downregulate cFLIP expression.157 Recently, it has been shown that administration of methyl-2-cyano-3, 12-dioxooleana-1, 9-dien-28-oate (CDDO-Me), a novel synthetic tri-terpenoid to human lung cancer cells, triggers induction of apoptosis by targeting cFLIPs to ubiquitination dependent, proteosomal degradation.159 siRNA mediated knock down of cFLIP potentiated the anti-cancer effect of CDDO-Me. CDDO-Me entered phase 1 clinical trial with a very promising results.159
Inhibitor of apoptosis proteins in cancer therapy
The inhibitor of apoptosis proteins (IAPs) were originally discovered in baculovirus as suppressors of host cell apoptosis160; however, they can be found in both invertebrates and vertebrates. Thus far, eight human IAPs family members have been identified including neuronal apoptosis inhibitory protein or (NAIP) (also known as BIRC1 or baculoviral IAP repeat-containing 1), cellular IAP 1 or cIAP1 (also known as HIAP2, MIHB, and BIRC2), cIAP2 (also known as HIAP1, MIHC, and BIRC3), X-chromosome linked IAP or XIAP (also known as hILP, MIHA, and BIRC4), survivin (also known as TIAP and BIRC5), Apollon (also known as Bruce and BIRC6), melanoma IAP or ML-IAP (also known as KIAP, livin, and BIRC7), and IAP-like protein 2 (also known as BIRC8), which are reviewed elsewhere.161
All IAP proteins share two to three common structures of baculovirus IAP repeat (BIR) domains to bind and inactivate caspases, except survivin, the smallest human IAP protein which contains only a single BIR repeat.161 Most of the IAP proteins, excluding survivin, possess a carboxyl-terminal RING domain containing ubiquitin ligases required for ubiquitination and proteasomal degradation of caspases.161 XIAP is the most efficient caspase inhibitor among the IAP family members.162 Inhibition of apoptosis by XIAP is mainly coordinated through binding to initiator caspase-9 and effector caspases-3 and -7.163
Elevated expression of IAPs in several human malignancies has been reported. Tamm et al164 investigated expression of IAPs in 60 human tumour cell lines at mRNA and protein levels and found higher expression of XIAP and cIAP1 in most cancer cell lines analysed. Elevated expression of IAP family members in malignant cells can be influenced by different intra- and/or extracellular factors such as TNF. Survivin, an atypical IAP, is highly expressed in rapidly dividing cells and many cancers.165 166 Espinosa and colleagues167 investigated the expression of several IAPs including c-IAP1, cIAP2, XIAP and survivin in cervical cancer. Although their finding indicates no differences in the expression of cIAP2 and XIAP between normal vs cancer samples, higher expression of survivin isoforms, 2B and DeltaEx3, along with downregulation of cIAP1 were detected in the cervical cancer samples. Nuclear expression of survivin in cancer cells has also been reported by Giodini and colleagues,168 demonstrating its role in cell division via controlling of microtubule stability and assembly of a normal mitotic spindle. Authors also hypothesised that nuclear localisation of survivin in cancer cells may facilitate checkpoint evasion and promote resistance to drugs targeting mitotic spindles.168
Attempts have been made to therapeutically target IAP proteins by small molecules or by antisense approaches.169 For example, OSU-03012 is a potent experimental anticancer drug, a derivative of celecoxib, known for its activity on multiple myeloma cells.170 One of its proposed mechanisms of action is downregulation of the expression of inhibitor for caspases such as survivin and XIAP, followed by cell cycle arrest and induction of apoptosis.
Triptolide, a diterpenoid isolated from a Chinese herb, induces a broad range of anticancer activities on solid tumours.171 For example, triptolide induced caspase dependent apoptosis by downregulating XIAP, and enhancing mitochondrial death pathway by activating caspase-9. A recent report published by Carter and colleagues demonstrates that triptolide can also enhance TRAIL induced apoptosis in acute myeloid leukaemia (AML) cells by downregulating the expression of XIAP and elevating the levels of DR5, a receptor for TRAIL.172
cIAP1 protein level could be downregulated by a small molecule inhibitor, (-)-N- [(2S, 3R)-3-amino-2-hydroxy-4-phenyl-butyryl]-l-leucine methyl ester (ME-BS), that induces its auto-ubiqitilation.173 ME-BS directly interacts with BIR3 domain of cIAP1 and promotes its proteosomal degradation.
Another approach enhancing apoptosis is based on over-expression of Smac/DIABLO, an IAP-inhibitor, and subsequent treatment with anticancer drugs such as doxorubicin, etoposide, paclitaxel and tamoxifen. Such combined treatment increased the total number of apoptotic breast cancer cells as compared with using the respective anticancer drugs alone.174 Smac/DIABLO overexpression also sensitised breast cancer cells to TRAIL. Several attempts have been made to develop cell permeable N-terminal peptides derived from the Smac that would serve itself as IAP inhibitors.175 176 Co-administration of these Smac peptides with etoposide, doxorubicin, and TRAIL resulted in alleviation of IAP effects as well as an increased apoptotic response. This was observed in several tumour cell lines, including breast, neuroblastoma, melanoma, and NSCLC as well as in a malignant glioma xenograft model in vivo.176 177 Like other peptides, Smac peptides might be difficult to use in clinical settings due to stability issues, immunogenicity and poor tissue distribution. Thus, the next generation of such drugs will likely be based on peptidomimetics.
AEG35156 is a 19-mer oligonucleotide that is being developed by Aegera therapeutics (Montreal, Canada). AEG35156 efficiently reduced the mRNA level of XIAP and sensitised cancer cells to apoptotic cell death. It also exhibited potent anti-tumour activity in the human cancer xenograft models.178 Presently, the drug is in phase 1 clinical trial in cancer patients as a single agent as well as in combination with docetaxel.178
N-[(5R)-6-[(anilinocarbonyl) amino]-5-((anilinocarbonyl), amino) hexyl]-N-methyl-N’-phenylurea (1396–12), one of the antagonists of XIAP, a small molecule inhibitor and a member of polyphenyl urea, efficiently induced apoptosis in AML cells and in the primary patient samples by downregulating expression of XIAP.179 However it did not have any cytotoxic effects on the normal haematopoietic cells, indicating its specificity in killing cancer cells.179 Other XIAP small molecule antagonists, such as 1396–11 and 1396–28, have been developed.180 In vitro studies using these compounds in pancreatic cancer cell lines and also in xenograft models showed that they can impede neoplastic growth by inhibiting XIAP. These compounds also showed synergy when applied in combination with TRAIL, gemcitabine, and radiation.180
Survivin has been recently singled out among the factors responsible for the resistance of colorectal cancer to standard therapies.181 Using siRNA technology, it has been demonstrated that inhibition of survivin potentiated the cancer cell death upon irradiation.181 Interaction of survivin with hsp90 (heat shock protein 90) is well documented and this interaction is vital for the stability of surviving.182 A cell permeable peptidomimetic, shephedrin, is based on the binding interface of survivin and Hsp90.183 Shephedrin destabilises the above interaction, thus depleting survivin, and selectively induces apoptosis only in tumour cells but not in normal cells, by both caspase dependent and independent mechanisms.183 shRNA mediated inhibition of survivin expression combined with the introduction of apoptin, a viral protein that selectively kills cancer cells, has recently been tested, and the data reveal synergistic effects of both treatments.184 185 Apart from the above approaches, anti-survivin hammerhead ribozymes have been developed that can effectively downregulate survivin’s expression, and hence potentiate cancer cell apoptosis.186–188
Direct activation of caspases by pharmacologic agents
Numerous approaches to trigger direct caspase activation specifically in tumour cells have been tested. This tactic appears to be very promising since some caspases, most prominently procaspase-3, are maintained in an inactive conformation by an Asp-Asp-Asp “safety-catch”, a regulatory tripeptide located within a flexible loop near the large subunit/small subunit junction. When these interactions are disturbed by mutation or by simple pH lowering, a substantial proportion of procaspase-3 molecules undergo spontaneous auto-activation.
Following this approach, Jiang and colleagues identified a small molecule drug, α-(trichloromethyl)-4-pyridineethanol (PETCM), that could activate procaspase-3 in cell extracts.189 However, this compound is an unlikely therapeutic agent, as high concentrations (200 μM) are required to activate caspase-3 in vitro. A series of small molecule caspase-activating drugs (MX-2060), derivatives of gambogic acid, were also evaluated for direct caspase-3 activation.190 Gambogic acid’s relatively low half maximal effective concentration (EC50) of 0.78 μM in a caspase activation assay in T47D breast cancer cells makes it a more attractive candidate for a caspase activating drug. Its derivative, MX-2167, has been shown to induce apoptosis in prostate, breast, colorectal and lung cancer cell lines, and to suppress tumour growth up to 90% in a syngeneic prostate animal cancer model.
Most recently, a small molecule, PAC-1, has been identified that directly activates procaspase-3 in vitro (EC50 for activation of 0.22 μM on procaspase-3) and induces apoptosis in tumour cells isolated from primary colon cancer in a manner directly proportional to the concentration of procaspase-3 inside these cells.191 This compound prevented tumour growth in three different murine models of cancer, including two models in which PAC-1 was administered orally.191 In fact, a systematic evaluation of procaspase-3 concentrations in the panel of 60 cell lines used by the National Cancer Institute revealed that particular lung, melanoma, renal and breast cancers show greatly enhanced concentrations of procaspase-3.192 Therefore, targeting caspase-3 activation could be a valuable strategy for cancer therapy.
PAC-1 induces apoptosis in a variety of cancer cell lines. In HL-60 cells, the addition of PAC-1 induced the appearance of many apoptotic hallmarks.191 PAC-1 treatment cause considerable phosphatidylserine externalisation as assessed by annexin V staining, a hallmark of apoptosis. This effect was observed at PAC-1 concentrations between 5–100 μM. PAC-1 also induced chromatin condensation in HL-60 cells, as visualised by Hoechst-33258 staining.191 Furthermore, PAC-1 mediated caspase-3 activation was confirmed by detection of cleaved caspase substrate poly-ADP-ribose polymerase 1 (PARP-1), and observed mitochondrial membrane depolarisation.191 Perhaps most interestingly, PAC-1 shows some selective toxicity towards cancer cells. When tested on matched normal epithelial and colon adenocarcinoma cells obtained from the same donors, the IC50 values for cancer cells were between 0.003–1.41 μM, whereas for normal cells the IC50 values were 5.02–9.98 μM. The increased susceptibility of colon cancer cells to PAC-1 correlated well with 1.7–19.7-fold (average of 8.4-fold) increased expression of procaspase-3 in cancer cells as compared to their normal counterparts.191
EPILOGUE
The apoptotic potential of cancer cells in correlation to their proliferative dynamics profoundly affects malignant phenotypes, and it appears that pathways governing cell proliferation and cell death are interconnected.193 194 Failure to enter apoptosis allows transformed cells to enter further cell divisions and acquire further mutations. In the present review, we focus on genetic alterations of caspases and their regulators, underlining the role of these molecules in cancer development. Deregulation of caspase expression and/or activity could be a result of various factors, including genetic alterations, promoter methylation, alternative splicing and posttranslational modifications.139 195–197 We show examples that different mutation could have profound effects on caspases activity.
The majority of currently available anticancer drugs act at least in part through induction of apoptosis198–200; therefore, a defect in the apoptotic propensity of tumours affects their response to treatment. Some experimental treatments—for example, apoptin—seem to “hijack” cell’s proliferation promoting pathways, and redirect them to induce apoptosis.201 202 Beside traditional radio- and chemotherapies, new treatment methods are being developed that utilise natural products such as Brevinin-2R, and immunomodulators such as S100A8/A9, and even utilisation of stem cells.20 24 203 204 As described above, a number of anticancer therapies are being tested that influence the expression and/or activity of factors that regulate apoptosis. Targeting caspases and apoptotic machinery will play an increasingly important role in future modern cancer therapy, and approaches are being developed that allow “on demand” activation of expression.205 This will be achieved using siRNA technology, the small molecule inhibitors, as well as peptides and peptidomimetics. These approaches may eventually replace the traditional chemo- and radiation therapies, and result in more efficient cancer treatments that are devoid of side effects.
Acknowledgments
ML acknowledges the support from DFG (SFB 773, GRK 1302) and the Deutsche Krebshilfe. We also acknowledge and apologise to all those authors whose work was not directly referenced here due to space limitations.
REFERENCES
Footnotes
Funding: SG is supported by a CIHR/Canadian Lung Association/GSK Fellowship, Manitoba Institute of child Health (MICH), and by the National Training Program in Allergy and Asthma (NTPAA). AJH holds a Canada Research Chair; ME is supported by a MICH studentship, and BY is supported by CIHR, Manitoba Health Research Council (MHRC), and Institute of Cardiovascular Sciences (ICS) studentship.
Competing interests: None declared.