Article Text

Abstract
Congenital hyperinsulinism (CHI) is biochemically characterised by the dysregulated secretion of insulin from pancreatic β-cells. It is a major cause of persistent hyperinsulinaemic hypoglycaemia (HH) in the newborn and infancy period. Genetically CHI is a heterogeneous condition with mutations in seven different genes described. The genetic basis of CHI involves defects in key genes which regulate insulin secretion from β-cells. Recessive inactivating mutations in ABCC8 and KCNJ11 (which encode the two subunits of the adenosine triphosphate sensitive potassium channels (ATP sensitive KATP channels)) in β-cells are the most common cause of CHI. The other recessive form of CHI is due to mutations in HADH (encoding for-3-hydroxyacyl-coenzyme A dehydrogenase). Dominant forms of CHI are due to inactivating mutations in ABCC8 and KCNJ11, and activating mutations in GLUD1 (encoding glutamate dehydrogenase) and GCK (encoding glucokinase). Recently dominant mutations in HNF4A (encoding hepatocyte nuclear factor 4α) and SLC16A1 (encoding monocarboxylate transporter 1) have been described which lead to HH. Mutations in all these genes account for about 50% of the known causes of CHI. Histologically there are three (possibly others which have not been characterised yet) major subtypes of CHI: diffuse, focal and atypical forms. The diffuse form is inherited in an autosomal recessive (or dominant manner), the focal form being sporadic in inheritance. The diffuse form of the disease may require a near total pancreatectomy whereas the focal form requires a limited pancreatectomy potentially curing the patient. Understanding the genetic basis of CHI has not only provided novel insights into β-cell physiology but also aided in patient management and genetic counselling.
Statistics from Altmetric.com
Congenital hyperinsulinism (CHI) is biochemically characterised by the dysregulated secretion of insulin from pancreatic β-cells. It is a major cause of hypoglycaemia in the newborn and infancy period.1 The unregulated insulin secretion drives glucose into insulin sensitive tissues (skeletal muscle, liver and adipose tissue) and prevents the generation of alternative energy substrates (such as ketone bodies), thus depriving the brain of both its primary and secondary energy sources.2 This prevailing metabolic milieu is the main factor leading to hypoglycaemic brain injury and mental retardation in this group of patients.
The incidence of CHI can vary from 1 in 40 000–50 000 in the general population to 1 in 2500 in some isolated communities with high rates of consanguinity.3–5 It is a heterogeneous condition with regards to the clinical presentation, the underlying genetic aetiology, molecular mechanisms and histological basis of the disease.6–9 The clinical presentation can be heterogeneous ranging from completely asymptomatic, mild medically responsive to severe medically unresponsive disease which may require a near total pancreatectomy. Those patients undergoing a total pancreatectomy have a high risk of developing post-pancreatectomy diabetes mellitus and pancreatic exocrine insufficiency.
Insulin secretion from β-cells is precisely regulated to maintain blood glucose values within the normal range (3.5–5.5 mmol/l). The genetic basis of CHI involves defects in key genes which regulate insulin secretion from β-cells. The most common cause of CHI are recessive inactivating mutations in ABCC8 and KCNJ11 which encode the two subunits of the adenosine triphosphate sensitive potassium channels (ATP sensitive KATP channels) in the pancreatic β-cell.10–17 These β-cell KATP channels play a key role in transducing signals derived from glucose metabolism to β-cell membrane depolarisation and regulated insulin secretion.18 The other rare recessive form of CHI is due to mutations in HADH (encoding for-3-hydroxyacyl-coenzyme A dehydrogenase).19–21 Dominant forms of CHI are due to inactivating mutations in ABCC8 and KCNJ1122–25 and activating mutations in GLUD1 (encoding glutamate dehydrogenase),26–31 GCK (encoding glucokinase),32–38 HNF4A (encoding hepatocyte nuclear factor 4α)39–41 and SLC16A1 (encoding monocarboxylate transporter 1).42 Mutations in all these genes account for about 50% of the known causes of CHI, and in some populations mutations in these genes account for only about 20% of CHI cases,5 9 43 44 suggesting other novel genetic aetiologies.
Histologically there are three major subgroups of the disease: diffuse, focal, and atypical.45 46 The diffuse forms of CHI are inherited in an autosomal recessive or dominant manner and the focal form is sporadic in inheritance. The diffuse form of the disease may require a near total pancreatectomy (with a high risk of developing diabetes mellitus) whereas the focal form requires a limited pancreatectomy offering a complete “cure” for the patient. In patients with atypical disease the histological abnormalities are either more extensive while remaining limited, or diffuse with the coexistence of normal and abnormal islets.46
Understanding the genetic basis of CHI has not only provided novel insights into β-cell physiology but also aided in patient management and genetic counselling. In terms of patient management rapid genetic analysis for mutations in ABCC8 and KCNJ11 can help in the genetic diagnosis of diffuse or focal CHI.47 Prenatal diagnosis of CHI based on the genetic analysis of known family members with mutations in ABCC8 (and KCNJ11) is also now possible permitting immediate medical management at the time of birth.48 This state of the art review will firstly give a brief overview of the role of β-cell KATP channels in regulating insulin secretion, and then focus in detail on the genetic and molecular basis of CHI due to mutations in the known genes and highlight some of the more recent genetic advances. Table 1 summarises the known genetic causes of CHI.
THE ROLE OF KATP CHANNELS IN REGULATING GLUCOSE INDUCED INSULIN SECRETION
KATP channels have a key role in the physiology of many cells, and defects either in the channel itself or in its regulation can lead to diseases in humans.49 50 Functionally KATP channels provide a means of linking the electrical activity of a cell to its metabolic state by sensing changes in the concentration of intracellular nucleotides, and in some cases they mediate the actions of hormones and transmitters.51 The pancreatic KATP channel is a functional complex of the sulfonylurea receptor 1 (SUR1) and an inward rectifier potassium channel subunit (Kir6.2) and plays a pivotal role in regulating insulin secretion from the β-cell.52 The Kir6.2 forms the pore of the channel and the SUR1 (an ATP binding cassette transporter) acts as a regulatory subunit.
KATP channels are regulated by adenine nucleotides to convert changes in cellular metabolic levels into membrane excitability. Each subunit of the KATP channel is known to be differentially regulated. The Kir6.2 subunit determines the biophysical properties of the channel complex including K+ selectivity, rectification, inhibition by ATP and activation by acyl-CoAs.53 The sulfonylurea receptors endow KATP channels with sensitivity to the stimulatory actions of Mg-nucleotides and KATP channel openers (for example, diazoxide, nicorandil) and the inhibitory effects of sulfonylureas and glinides54 and endosulfins.55
The molecular topology of SUR1 consists of three transmembrane domains, TMD0, TMD1, andTMD2, each of which consists of five, five, and six membrane spanning regions, respectively.56 SUR1 also has two nucleotide binding folds (NBF-1 and NBF-2) on the cytoplasmic side with which it senses changes in intracellular [ATP]/[ADP] and transmits the signal to the pore. NBF1 appears to be the principal site for ATP binding, whereas NBF2 binds MgADP.56 NBF-1 and NBF-2 are located in the loop between TMD1 and TMD2 and in the C-terminus, respectively. These binding domains cooperate with each other in mediating the nucleotide regulation of the pore function.57 NBFs of SUR contain highly conserved motifs among ABC proteins: Walker A motif, Walker B motif, ABC signature motif (also called linker sequence or LSGGQ motif), and an invariant glutamine and histidine residue (also called the Q-loop and H-loop, respectively). Walker A and Walker B motifs are directly involved in nucleotide binding.58
KATP channels can only function if they are assembled and correctly transported to the cell membrane surface (trafficking). The assembly and trafficking of KATP channels are intricately linked processes. Only octameric KATP channel complexes are capable of expressing on the cell membrane surface. For example both Kir6.2 and SUR1 possess an endoplasmic reticulum (ER) retention signal (RKR) that prevents the trafficking of each subunit to the plasma membrane in the absence of the other subunit.59 Correct assembly of the two subunits masks these retention signals, allowing them to traffic to the plasma membrane. The retention signal is present in the C-terminal region of Kir6.2 and in an intracellular loop between TM11 and NBF-1 in SUR1. Truncation of the C-terminus of Kir6.2 deletes its retention signal, allowing functional expression of Kir6.2 in the absence of SUR1 subunit.60 In addition to these retrograde signals, the C-terminus of SUR1 has an anterograde signal, composed in part of a dileucine motif and downstream phenylalanine, which is required for KATP channels to exit the ER/cis-Golgi compartments and transit to the cell surface.61 Deletion of as few as seven amino acids, including the phenylalanine, from SUR1 markedly reduces surface expression of KATP channels.62 Thus, one function of SUR is as a chaperone protein, to facilitate the surface expression of Kir6.2. There is also some evidence that Kir6.2 provides a reciprocal service for SUR.63
The SUR1 protein shows a high affinity binding capacity to the sulfonylurea glibenclamide, indicating that SUR1 confers sulfonylurea binding.64 65 The sulfonylurea drugs (glibenclamide and tolbutamide) inhibit the channels and are used in the treatment of non-insulin dependent (type II) diabetes mellitus. The other class of drugs, known as potassium channel openers (for example, diazoxide), activate the channel and are used to suppress insulin secretion.65
GENETIC BASIS OF CHI DUE TO RECESSIVE INACTIVATING MUTATIONS IN ABCC8 AND KCNJ11
The ABCC8 gene consists of 39 exons and spans more than 100 kb of genomic DNA.66 The human SUR1 cDNA contains a single open reading frame that encodes for 1582 amino acids with a molecular weight of 177 kDa (GenBank NM_000352.2). Kir6.2 consists of a single exon encoding for a protein of 390 amino acids (GenBank NM_000525.2).52 The ABCC8 and KCNJ11 genes are both located on chromosome 11p15.1, separated by only a small stretch of 4.5 kb of DNA.66 Homozygous, compound heterozygous and heterozygous recessive inactivating mutations (missense, frameshift, nonsense, insertions/deletions (macrodeletion), splice site and regulatory mutations) have been reported in ABCC8 and KCNJ11.10–17 67 68 So far, more than 150 mutations have been reported in ABCC8 and 25 in KCNJ11.69 In the Ashkenazi Jewish population two common mutations (F1388del and c.3992–9G4A) account for 90% of all cases of CHI9 10 whereas in the Finnish population, two founder mutations have been reported (V187D and E1507 K).14 22 Recessive inactivating mutations in ABCC8 and KCNJ11 usually cause severe CHI which in the vast majority of patients is unresponsive to medical treatment with diazoxide. However, some compound heterozygote mutations may be milder and may respond to treatment with diazoxide.70 Compound heterozygote mutations may result in complex interactions resulting in intracellular retention of channel complexes.71 The molecular basis of recessive inactivating ABCC8 and KCNJ11 mutations involves multiple defects in KATP channel biogenesis and turnover, in channel trafficking from the ER and Golgi apparatus to the plasma membrane and alterations of channels in response to both nucleotide regulation and open state frequency. Figure 1 shows a schematic outline of the β-cell KATP channel and locations of the some of the common mutations.

(A) Schematic outline of the components of the β-cell KATP channel. The KATP channel is composed of two proteins: SUR1 which consists of 17 transmembrane domains with two intracellular nucleotide binding (NBF) motifs. Two N-linked glycosylation sites are present on amino acids 10 and 1049. Kir6.2 has three transmembrane segments. Common mutations in both of the proteins are highlighted. (B) The hetero-octameric arrangement of the KATP channel.
Recessive ABCC8 and KCNJ11 mutations resulting in defects in channel biogenesis and turnover
The mechanisms that control the maturation and assembly of KATP channels are not well understood. Pulse labelling studies have shown that when Kir6.2 is expressed individually, its turnover is biphasic with about 60% being lost with a half life of 36 min.72 The remainder converts to a long lived species (half life 26 h) with an estimated half time of 1.2 h. Expressed alone SUR1 has a long half life of 25.5 h. When Kir6.2 and SUR1 are co-expressed, they associate rapidly and the fast degradation of Kir6.2 is eliminated.72 Two mutations, KCNJ11 (W91R) and ABCC8 (F1388del), identified in patients with the severe form of CHI, profoundly alter the rate of Kir6.2 and SUR1 turnover, respectively.72 Both mutant subunits associate with their respective partners but dissociate freely and degrade rapidly, suggesting that the mutations alter channel biogenesis and turnover.
Recessive inactivating mutations in ABCC8 and KCNJ11 resulting in defects in channel trafficking
Trafficking of KATP channels requires that the ER retention signal, RKR, present in both SUR1 and Kir6.2, is shielded during channel assembly. Some mutations in ABCC8 (such as R1437Q(23)X, F1388del and R1394H0) cause a trafficking defect by affecting the exit of channel subunits from the ER compartment.13 73–75 The R1437Q(23)X, mutation in exon 35 of ABCC8 truncates 200 amino acids from the COOH terminal region of the protein, an area that contains the anterograde signalling sequence (L1566, L1567.F1574) and residue L1544 which is part of the cloaking region for the RKR sequence.76 This defect affects the exit of channel subunits from the ER.
The pivotal role played by the RKR signal in allowing the channels to express correctly on the β-cell membrane is illustrated by the fact that when the RKR signal is inactivated by an in-frame deletion (F1388del SUR1AAA), channel activity is impaired, but when surface expression is rescued, the channels function normally.73 Other mutations such as the R1394H (ABCC8) cause a trafficking disorder by effecting retention of mutant proteins in the trans-Golgi network.74
Mutations in KCNJ11 can also cause defective trafficking and truncated non-functional proteins. For example, the Kir6.2 mutation (Y12X) causes the synthesis of a truncated non-functional protein12 whereas another mutation (W91R) leads to defective channel assembly with a rapid degradation in the ER.72 Recently, a new homozygous mutation H259R (KCNJ11) has been shown to lead to a non-functional KATP channels with impaired trafficking to the cell membrane.16
Recessive inactivating mutations in ABCC8 and KCNJ11 resulting in defects of channel regulation
The SUR1 subunit plays a key role in determining the pharmacological regulation of KATP channels with SUR1 acting as a conductance regulator of Kir6.2. The sensitivity of KATP channels to changes in ATP, ADP, and guanosine (GTP, GDP) nucleotides involves both subunits. The functional regulation of KATP channels is induced by changes in the ATP/ADP ratio. This involves cooperative interactions of nucleotides at both subunits with the actions of ATP induced inhibition of Kir6.2 being countered by the activation of ADP at SUR1. Hence, mutations that affect the regulation of the KATP channels by altering its sensitivity to changes in ADP/ATP will lead to unregulated insulin secretion. Several mutations in ABCC8 (for example, R1420C, T1139M and R1215Q) have now been described that result in the loss of ADP dependent gating properties of the channel.75 77 78 Loss of ADP dependent gating results in the constitutive inhibition of KATP channels by ATP.
Dominant inactivating ABCC8 and KCNJ11 mutations causing CHI
Dominant inactivating mutations in ABCC8 and KCNJ11 have been described which lead to CHI.22–25 However, the phenotype of patients with dominant inactivating mutations in ABCC8 and KCNJ11 seems to be much milder than that of patients with recessive inactivating ABCC8 and KCNJ11 mutations. Patients with dominant mutations seem to be responsive to medical treatment with diazoxide, may present later than those with recessive mutations, and do not require a pancreatectomy.24 In one large study of 16 families with dominant CHI caused by mutations in ABCC8 or KCNJ11, all of the mutations were conservative single amino acid changes, allowing for normal channel formation at the plasma membrane.24 Whereas recessive mutations cause near absence of KATP channel activity (to have defects in channel biogenesis or trafficking of mature functional channels to the plasma membrane), dominant mutations demonstrate normal channel assembly with their respective wild type partner and normal trafficking of assembled channels to the plasma membrane when expressed in vitro.
A dominant missense CHI causing mutation F55L (KCNJ11) has been shown to greatly reduce the open probability of KATP channels in intact cells without affecting channel expression.25 It was shown that the low channel activity was due to reduced channel response to membrane phosphoinositides and/or long chain acyl-CoAs, as application of exogenous PIP2 or oleoyl-CoA restored channel activity similar to that seen in wild type channels. These electrophysiological observations provide a link between KATP channels and their regulation by membrane phosphoinositides and/or long-chain acyl-CoAs.79
DOMINANT ACTIVATING MUTATIONS IN GLUD1
The GLUD1 gene is located on chromosome 10q23.3 and contains 13 exons coding for a 505 amino acid mature enzyme, glutamate dehydrogenase (GDH).80 GDH is a mitochondrial matrix enzyme which is expressed at high levels in the liver, brain, kidney, pancreas, heart and lungs.81 This enzyme catalyses the oxidative deamination of glutamate to α-ketoglutarate and ammonia using NAD+ and/or NADP+ as co-factors. In the β-cell α-ketoglutarate enters the tricarboxylic acid cycle and leads to an increase in the cellular ATP. This increases the ATP/ADP ratio which triggers closure of the KATP channels and depolarisation of the β-cell membrane. This, in turn, opens the voltage gated calcium channel, raises the cytosolic calcium, and triggers the release of insulin. GDH plays a critical step in glutaminolysis and regulating amino acid induced insulin secretion.82 Its activity is regulated by a complex interplay of allosteric activators and inhibitors. Positive allosteric effectors of GDH include leucine, and ADP (adenine diphosphate), whereas GTP (guanosine 5′-triphosphate) is a potent allosteric inhibitor.83 Allosteric activation of glutaminolysis is one mechanism by which the amino acid leucine stimulates insulin secretion.83
Activating mutations (heterozygous missense single amino acid substitutions) in the GLUD1 gene are the second most common cause of CHI. GLUD1 gene mutations cause a form of CHI in which affected children have recurrent symptomatic HH together with a persistently elevated plasma ammonia value, the hyperinsulinism/hyperammonaemia (HI/HA) syndrome.26 84–86 The mutations causing HI/HA reduce the sensitivity of the enzyme to allosteric inhibition by the high energy phosphate GTP27 86 and in rare cases increase basal GDH activity.28 29 87 The loss of inhibition by GTP increases the rate of oxidation of glutamate in the presence of leucine, thereby increasing insulin secretion. The clinical picture is hence characterised by postprandial hypoglycaemia following a protein meal (fasting hypoglycaemia may also occur). The mechanism of persistent hyperammonaemia,86 88 a striking and consistent feature of this condition, is not completely understood. The hypoglycaemia in patients with HI/HA syndrome is usually responsive to medical treatment with diazoxide. The hyperammonaemia is considered to be asymptomatic and hence efforts to reduce plasma ammonia values with sodium benzoate or N-carbamylglutamate do not seem to be beneficial.
Glutamate dehydrogenase is a homohexameric enzyme with two trimeric subunits; each subunit is composed of at least three domains. Mutations in GLUD1 occur most commonly in the GTP allosteric binding domain of GDH (exons 11 and 12).26 27 Mutations in the catalytic domain (exons 6 and 7) have also been identified.29 30 This domain interacts with GTP molecules and mutations in the allosteric and catalytic domains have been shown to cause hyperinsulinism by diminishing the sensitivity of GDH to GTP. The third domain is an antenna-like structure connecting to the pivot helix where mutations (exon 10) have also been reported.28 31 Functional analysis of these mutations has shown that they are associated with a higher basal GDH activity and a milder insensitivity to GTP inhibition in comparison with mutations in the other two domains.28 31 Most cases of HI/HA syndrome occur sporadically; however, families with HI/HA syndrome where the mutation has been dominantly inherited have also been described.30 Figure 2 summarises the mutations in the GLUD1 gene known to cause CHI.

Schematic representation of the mutations in the GLUD1 gene known to cause congenital hyperinsulinism. Mutations are described to occur in three domains: the allosteric binding domain (exons 11 and 12), the catalytic domain (exons 6 and 7), and the antenna region (exon 10).
DOMINANT ACTIVATING MUTATIONS IN GCK
The glucokinase gene (GCK) is located on chromosome 7p15.3-p15.1 and comprises 12 exons which span ∼45,168 bp and encode for a 465 amino acid protein with a molecular weight of 52 191 Da. The gene is transcribed in various tissues but it has tissue specific promoters and is especially expressed in the pancreas, liver, and brain.89 The presence of tissue specific promoters allows differential regulation and transcription of different transcripts giving rise to three different sized versions of exon 1 (a, b, and c). In the pancreas the upstream promoter is functional, while in the liver the downstream promoter is used.89 Exon 1a is expressed in the pancreatic β-cells whereas exons 1b and 1c are expressed in the liver.89
Glucokinase (hexokinase IV or D) is one member of the hexokinase family of enzymes. The name, glucokinase, is derived from its relative specificity for glucose under physiologic conditions. Glucokinase is a key regulatory enzyme in the pancreatic β-cells. It operates as a monomer and phosphorylates glucose on carbon 6 with MgATP as a second substrate to form glucose-6-phosphate (G6P) as a first step in the glycolytic pathway. It plays a crucial role in the regulation of insulin secretion and has been termed the pancreatic β-cell sensor on account of its kinetics, because the rate of phosphorylation of glucose in the pancreatic β-cells is directly related to the concentration of glucose over a range of physiological glucose concentrations (4–15 mmol/l).90 These kinetic characteristics are the enzyme’s low affinity for glucose (concentration of glucose at which the enzyme is half maximally activated, S0.5, 8–10 mmol/l), cooperativeness with glucose (Hill number of ∼1.7), and lack of inhibition by its product G6P.90 The enzyme has at least two clefts, one for the active site, binding glucose and MgATP, and the other for a putative allosteric activator. Glucokinase activity is closely linked to the KATP and calcium channels of the β-cell membrane, resulting in a threshold for glucose stimulated insulin release of approximately 5 mmol/l, which is the set point of glucose homoeostasis.91
Heterozygous inactivating mutations in GCK cause maturity onset diabetes of the young (MODY), homozygous inactivating in GCK mutations result in permanent neonatal diabetes, whereas heterozygous activating GCK mutations cause CHI. So far seven activating GCK mutations (V455M, A456V, Y214C, T65I, W99R, G68V, S64Y) have been described that lead to CHI (fig 3).32–38 Activating GCK mutations increase the affinity of GCK for glucose and alter (reset) the threshold for glucose stimulated insulin secretion. All reported activating mutations cluster in a region of the enzyme, which has been termed the allosteric activator site and is remote to the substrate binding site. The allosteric site of GCK is where small molecule activators bind, suggesting a critical role of the allosteric site in the regulation of GCK activity.92 Both GCK activators and activating mutations increase enzyme activity by enhancing the affinity for glucose as described by a decrease in K0.5.93 There is no evidence to suggest that over-expression of GCK (increased gene dosage effect) is a likely cause of CHI.94
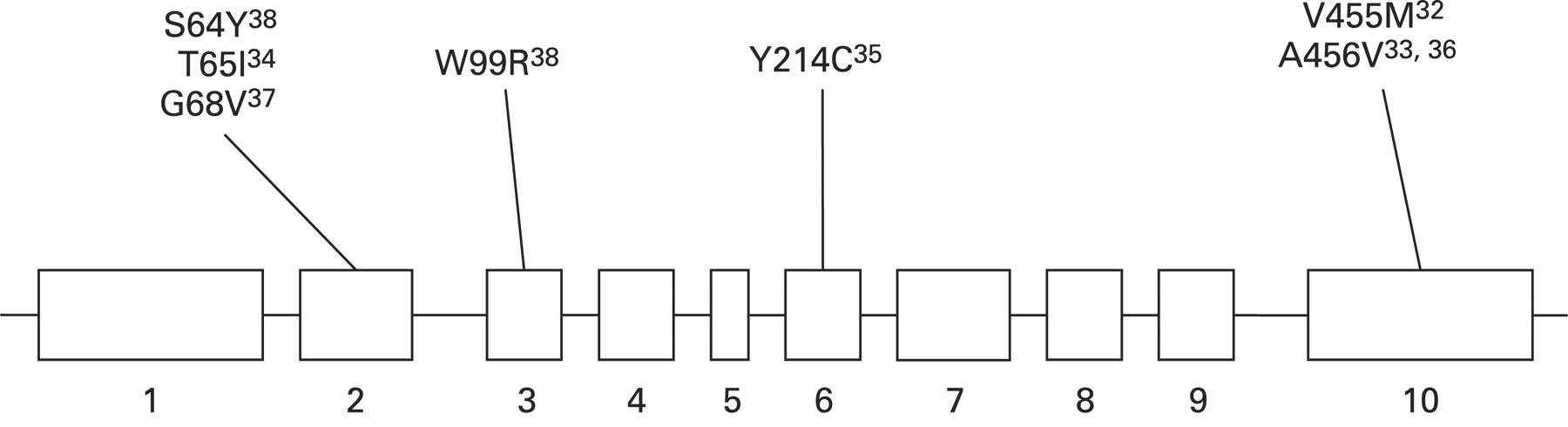
Schematic representation of the mutations in the GCK gene known to cause congenital hyperinsulinism.
The clinical symptoms and course of patients with GCK mutations cover a broad spectrum from asymptomatic hypoglycaemia to unconsciousness and seizures, even within the same family with the same mutation, implicating a complex mechanism for GCK regulation. Patients with activating GCK mutations may present with postprandial hyperinsulinaemic hypoglycaemia.95 Most of the GCK mutations reported to date cause mild diazoxide responsive CHI. However, a “de novo” mutation in GCK (Y214C) was described in a patient with medically unresponsive CHI.35 This mutation was located in the putative allosteric activator domain of the protein and functional studies of purified recombinant glutathionyl S transferase fusion protein of GK-Y214C showed a sixfold increase in its affinity for glucose, a lowered cooperativity, and increased kcat.35 The relative activity index of GKY214C was 130, and the threshold for glucose stimulated insulin secretion was predicted by mathematical modelling to be 0.8 mmol/l, as compared with 5 mmol/l in the wild-type enzyme.35 In the largest study performed to date on a pool of patients who were negative for mutations in the ABCC8 and KCNJ11 genes, the prevalence of CHI due to mutations in GCK was estimated to be about 7%.38
RECESSIVE MUTATIONS IN HADH
Mitochondrial fatty acid β-oxidation constitutes the essential physiological response to energy depletion caused by fasting, severe febrile illness or increased muscular activity. The process of β-oxidation results in production of acetyl-CoA by the sequential oxidation and cleavage of straight chain fatty acids. Importantly, hepatic β-oxidation provides the source of energy for extrahepatic tissues through ketone body formation upon fasting. HADH encodes for the enzyme L-3-hydroxyacyl-coenzyme A dehydrogenase (HADH) (previously known as short-chain L-3-hydroxyacyl-CoA dehydrogenase (SCHAD)), which is an intra-mitochondrial enzyme that catalyses the penultimate step in the β-oxidation of fatty acids, the NAD+ dependent dehydrogenation of 3-hydroxyacyl-CoA to the corresponding 3-ketoacyl-CoA. Human HADH encodes a 314 amino acid protein with eight exons and spans approximately 49 kb.96 It is composed of a 12 residue mitochondrial import signal peptide and a 302 residue mature HADH protein with a calculated molecular mass of 34.3kD.97
Loss-of-function mutations in the HADH gene are associated with CHI.19–21 The clinical presentation of all patients reported is heterogeneous, with either mild late onset intermittent HH or severe neonatal hypoglycaemia. All reported cases have presented with increased 3-hydroxyglutarate in urine and hydroxybutyrylcarnitine in blood which may be diagnostically useful markers for HADH deficiency. In the first patient reported sequencing of the HADH genomic DNA from the fibroblasts showed a homozygous mutation (C773T) changing proline to leucine at amino acid 258.19 Analysis of blood from the parents showed they were heterozygous for this mutation. Western blot studies showed undetectable levels of immunoreactive HADH protein in the patient’s fibroblasts. Expression studies showed that the P258L enzyme had no catalytic activity. This patient presented with intermittent hypoglycaemia at 4 months of age.
A novel, homozygous deletion mutation (deletion of the six base pairs CAGGTC at the start of HADH exon 5) was found in the second patient who presented with severe neonatal hypoglycaemia.21 The mutation affected RNA splicing and was predicted to lead to a protein lacking 30 amino acids. The observations at the molecular level were confirmed by demonstrating greatly reduced HADH activity in the patients’ fibroblasts and enhanced levels of 3-hydroxybutyryl-carnitine in their plasma. Urine metabolite analysis showed that HADH deficiency resulted in specific excretion of 3-hydroxyglutaric acid.21 Finally, the third patient reported to date was found to be homozygous for a splice site mutation (IVS6-2 a/g) in the HADH gene with western blotting with an anti-HADH antibody indicating a decrease in the amount of immunoreactive protein in fibroblasts from the patient consistent with the observed decrease in enzyme activity.20
The molecular mechanism of how loss of function in the HADH gene leads to unregulated insulin secretion is still unclear. Several recent studies in rodents have begun to give some insight into how HADH regulates insulin secretion and its interaction with other genes involved in β-cell development and function.98–101 The normal β-cell phenotype is characterised by a high expression of HADH and a low expression of other β-oxidation enzymes. Downregulation of HADH causes an elevated secretory activity suggesting that this enzyme protects against inappropriately high insulin values and hypoglycaemia.98 99 Hence, HADH seems to be a negative regulator of insulin secretion in β-cells. Further studies will be required to understand fully the biochemical pathways by which defects in HADH lead to dysregulated insulin secretion.
RECENT ADVANCES IN THE GENETIC AETIOLOGY OF CHI
A further mechanism of hyperinsulinaemic hypoglycaemia has been described whereby strenuous physical exercise causes an inappropriate burst of insulin release that can lead to hypoglycaemia.102 103 This work led to the identification of the molecular basis of exercise induced CHI (due to mutations in SLC16A1).42 Mutations in HNF4A have been recently reported to cause both transient and persistent hyperinsulinaemic hypoglycaemia associated with macrosomia and a family history of maturity onset diabetes of the young.39 41
DOMINANT MUTATIONS IN SLC16A1 CAUSING EXERCISE INDUCED CHI
In the glycolytic pathway glucose is metabolised to pyruvate which then enters in the mitochondria. Pyruvate can be converted into lactate or enters into the tricarboxylic acid cycle, generating reducing equivalents. This leads to stimulation of the respiratory chain and ATP synthesis. The transport of monocarboxylates such as lactate and pyruvate is mediated by the SLC16A family of proton linked membrane transport proteins known as monocarboxylate transporters (MCTs). Fourteen MCT related genes have been identified in mammals and of these seven MCTs have been functionally characterised. Despite their sequence homology, only MCT1–4 have been demonstrated to be proton dependent transporters of monocarboxylic acids.104
The SLC16A1 gene encodes for MCT1 that mediates the movement of lactate and pyruvate across cell membranes. The SLC16A1 gene maps to chromosome 1p13.2-p12, spans approximately 44 kb, and is organised as five exons intervened by four introns.105 106 The first of these introns is located in the 5′ UTR-encoding DNA, spans >26 kb, and thus accounts for approximately 60% of the entire transcription unit.106 Analysis of a 1.5 kb fragment of the MCT1 5′ flanking region shows an absence of the classical TATA-Box motif. However, the region contains potential binding sites for a variety of transcription factors.105
Studies in whole rat and mouse islets have shown that pyruvate and lactate cannot mimic the effect of glucose on insulin secretion despite active metabolism.107 108 This is postulated to be due to low expression of MCT in β-cells. However, over expression of lactate dehydrogenase (LDH) and MCT1 leads to pyruvate induced insulin secretion.108 In exercise induced CHI there is increased expression of MCT1 transporter in β-cells due to dominant mutations in SLC16A1.42 In these patients anaerobic physical exercise induces HH that is preceded by an inappropriate increase in the concentration of circulating insulin.102 103 Affected patients become hypoglycaemic within 30 min after a short period of anaerobic exercise. A pyruvate load test causes a brisk increase in the serum insulin concentration suggesting that pyruvate metabolism or transport is in some way involved in signalling insulin secretion from β-cells in these patients.109 A genome scan performed on two families with 10 affected patients first mapped the gene to chromosome 1.42 Mutations in the promoter region of SLC16A1 gene were confirmed in all patients. Studies on cultured fibroblasts from affected patients showed abnormally high SLC16A1 transcript levels, although the MCT1 transport activity was unchanged in fibroblasts (possibly reflecting additional post-transcriptional control of MCT1 levels in extrapancreatic tissues).
Two of the SLC16A1 mutations identified in separate pedigrees resulted in increased protein binding to the corresponding promoter elements and marked (3- or 10-fold) transcriptional stimulation of SLC16A1 promoter-reporter constructs.42 Some of the mutations were in the binding sites of several transcription factors (nuclear matrix protein 1, albumin negative factor (ANF) and AML-1a, simian-virus-40-protein-1 (Sp1), upstream stimulatory factor (USF), myeloid zinc finger 1 (MZF1), and GATA-1 binding site). These studies suggest that activating mutations in the promoter region of SLC16A1 could induce increased expression of MCT1 in the β-cell (where this gene is not usually transcribed) allowing pyruvate uptake and pyruvate stimulated insulin release despite ensuing hypoglycaemia.42
DOMINANT HETEROZYGOUS MUTATIONS IN HNF4A
Hepatocyte nuclear factor 4α (HNF4α) is a transcription factor of the nuclear hormone receptor superfamily and is expressed in liver, kidney, gut, and pancreatic islets.110 In combination with other hepatocyte nuclear factors, HNF4α has been proposed to form a functional regulatory loop that regulates the development and function of the pancreas and the liver.111 112 In β-cells, HNF4α has been shown to be the most widely acting transcription factor and regulates several key genes involved in glucose stimulated insulin secretion.113 114
The HNF4A gene is located on human chromosome 20q13.1–13.2. The gene consists of at least 12 exons and spans 30 kb. The HNF4A gene has two promoters, P1 and P2, with P2 being upstream to P1. The distant upstream P2 promoter represents the major transcription site in β-cells, and is also used in hepatic cells. Transfection assays with various deletions and mutants of the P2 promoter revealed functional binding sites for HNF1A, HNF1B, and IPF1.115 The HNF4α protein consists of an N-terminal ligand independent transactivation domain (amino acids 1–24), a DNA binding domain containing two zinc fingers (amino acids 51–117), and a large hydrophobic portion (amino acids 163–368) composed of the dimerisation, ligand binding, co-factor binding, and ligand dependent transactivation domain.116
Heterozygote mutations in the human HNF4A gene classically lead to maturity onset diabetes of the young subtype 1 (MODY1), which is characterised by autosomal dominant inheritance and impaired glucose stimulated insulin secretion from pancreatic β-cells.117 These mutations in the HNF4A gene cause multiple defects in glucose stimulated insulin secretion and in expression of HNF4A dependent genes.117 118 Recently mutations in the HNF4A gene were reported to cause macrosomia and both transient and persistent HH.39–41 In one retrospective study the birth weight of the HNF4A mutation carriers compared to non-mutation family members was increased by a median of 790 g.39 Transient hypoglycaemia was reported in 8/54 infants with heterozygous HNF4A mutations and documented HH in three cases.39
In another prospective study three infants presented with macrosomia and severe HH with a positive family history of MODY.41 All three patients required diazoxide treatment to maintain normoglycaemia. Sequencing of the HNF4A gene identified heterozygous mutations in all three families.41 In family 1, a frameshift mutation L330fsdel17ins9 (c.987 1003del17ins9; p.Leu330fs) was present in the proband; a mutation affecting the conserved A nucleotide of the intron 2 branch site (c.264-21A>G) was identified in the proband of family 2; and finally a nonsense mutation, Y16X (c.48C>G, p.Tyr16X), was found in the proband of family 3.41 Hence mutations in the HNF4A gene are a novel cause of both transient and persistent HH.
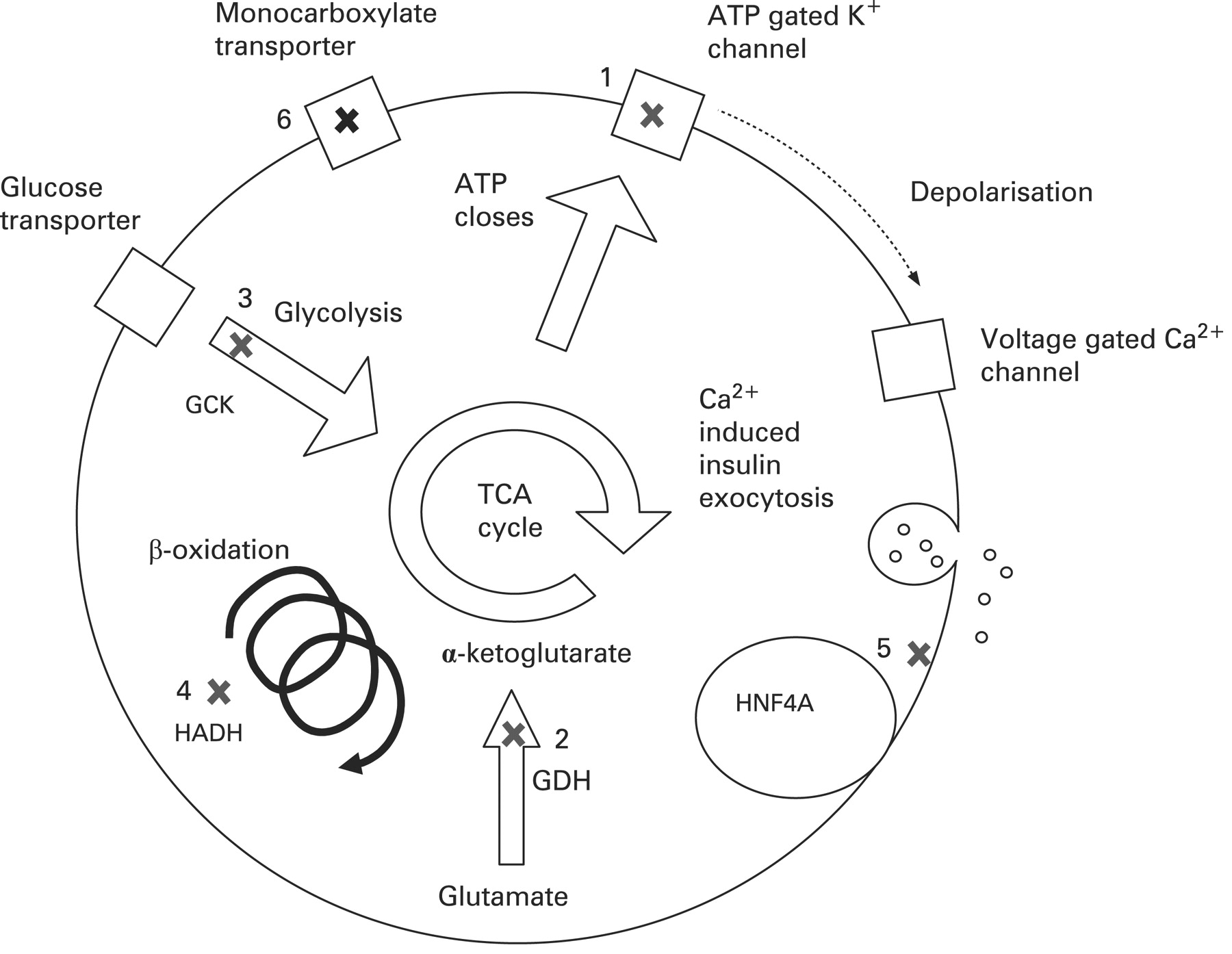
At present it is unclear how heterozygote mutations in HNF4A lead to hypoglycaemia. Using the conditional Cre-loxP-based inactivation system to delete HNF4A specifically in the pancreatic β-cell, Gupta et al studied glucose homeostasis in the adult mice.119 These mice were hyperinsulinaemic in fasted and fed state but also paradoxically had impaired glucose tolerance with inadequate insulin secretion after glucose stimulation. Cotransfection assays demonstrated that these mice had a 60% reduction in the expression of the Kir6.2 subunit of the potassium channel. However, two further studies39 120 have reported no change in the expression of Kir6.2 in HNF4A deficient mice suggesting that this loss of expression of Kir6.2 in the pancreatic β-cell may not be the cause of the dysregulated insulin secretion. Hence, it is unclear how heterozygous mutations in the HNF4A gene cause HH in the newborn period followed by the opposite phenotype of MODY-1 in young adulthood. Figure 4 summarises the genetic causes of CHI.

Summary of common mutations in the pancreatic β-cell leading to congenital hyperinsulinism. (1) The ATP gated potassium channel encoded by ABCC8 and KCNJ11. (2) Glutamate dehydrogenase (GDH) encoded by GLUD1. (3) Glucokinase (GCK), the initial enzyme in the glycolysis pathway. (4) L-3-hydroxyacyl-coenzyme A dehydrogenase (HADH), the penultimate enzyme in the β-oxidation pathway encoded by HADH. (5) Mutations in HNF4α cause multiple defects in glucose stimulated insulin secretion. (6) The monocarboxylate transporter MCT1 encoded by SLC16A1. TCA, tricarboxylic acid.
THE GENETIC BASIS OF FOCAL CHI
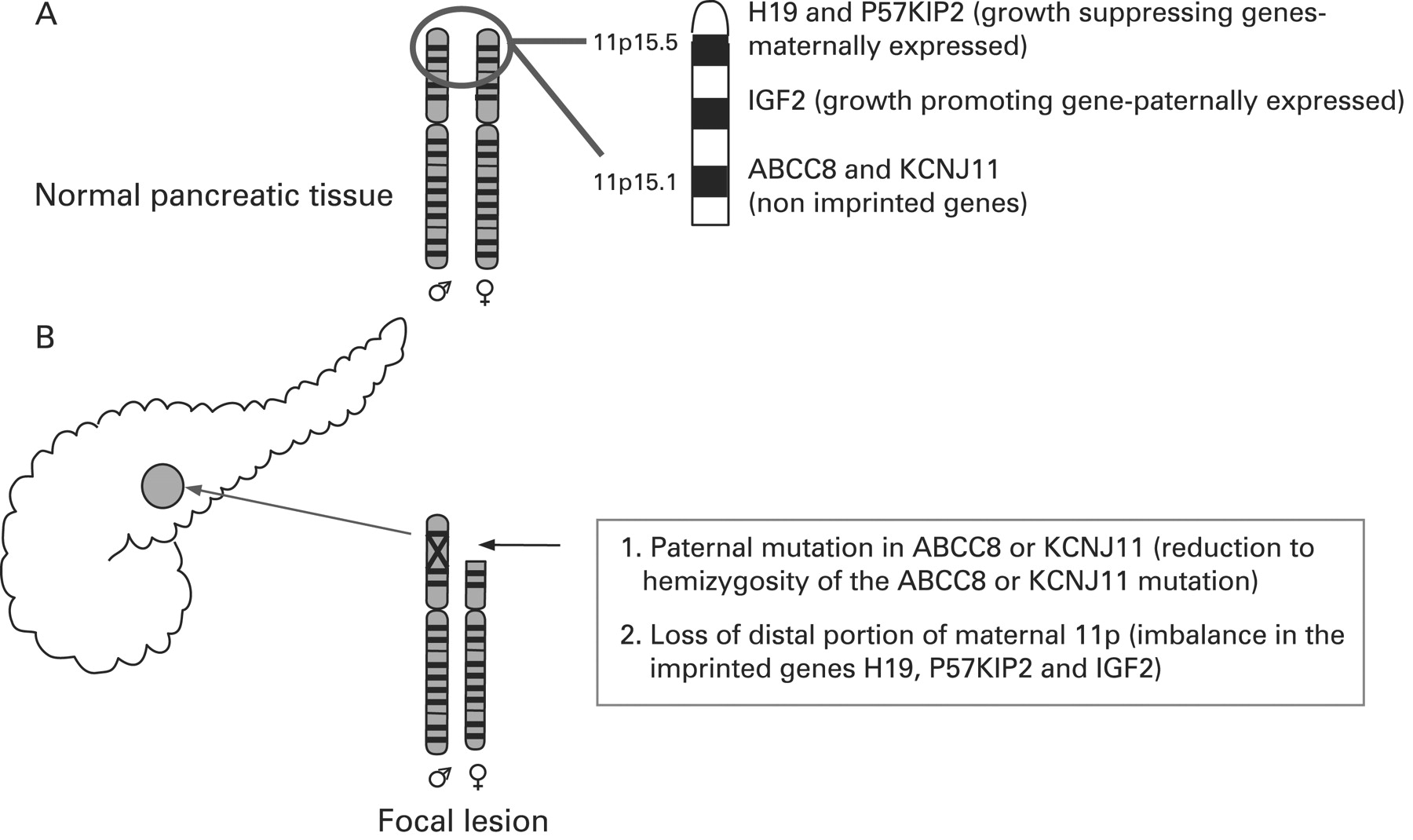
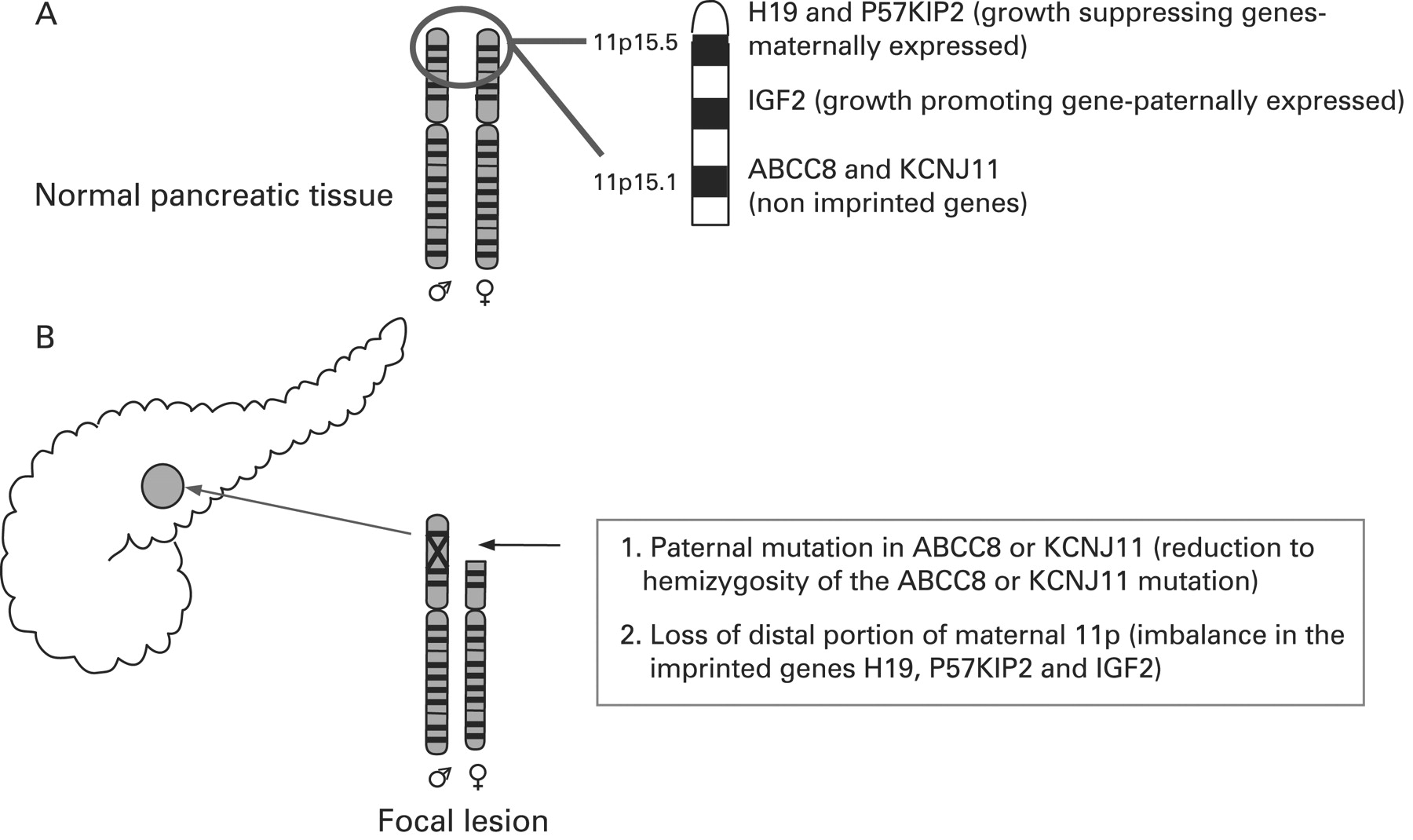
CHI presents as three (probably more, but these have not been fully defined yet) different morphological forms: a diffuse form with functional abnormality of islets throughout the pancreas; an atypical form where the pathophysiology is unclear; and a focal form with focal islet cell adenomatous hyperplasia, which can be cured by partial pancreatectomy.121 Focal CHI is characterised by nodular hyperplasia of islet-like cell clusters, including ductuloinsular complexes and giant β-cell nuclei.122 The genetic aetiology of focal CHI is distinct from that of diffuse CHI. Focal adenomatous hyperplasia involves the specific loss of the maternal 11p15 region and a constitutional mutation of a paternally inherited allele of the genes ABCC8/KCNJ11 encoding the KATP channel.123–128 The specific loss of the maternal 11p15 region within the focal region results in paternal isodisomy and a paternally inherited mutation in ABCC8/KCNJ11.124 129 The reduction to homozygosity of a paternally inherited ABCC8/KCNJ11 mutation within the focal lesion leads to uncontrolled secretion of insulin (fig 5).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Genetic aetiology of a focal lesion. Panel A shows normal parental chromosomes 11 with the distal region of the short arm containing the ABCC8 and KCNJ11 channel genes, and imprinted genes (H19 and P57KIP2 and IGF2), that influence cellular proliferation. Panel B explains the genetic basis of a focal lesion that results from paternal inheritance of a recessive ABCC8 or KCNJ11 mutation and the somatic loss of heterozygosity of the distal portion of the short arm of chromosome 11.
Deletion mapping experiments have shown that the most commonly deleted region encompasses two regions of interest: the 11p15.5 region, subject to imprinting, and the 11p15.4 region containing the ABCC8/KCNJ11 genes, which are not imprinted.123 The 11p15.5 chromosome region involved contains an imprinted domain, including several imprinted genes characterised by mono-allelic expression.130–133 These include four maternally expressed genes (H19, a candidate tumour suppressor gene; P57KIP2, a negative regulator of cell proliferation; KVLQT1, the gene coding for the potassium channel involved in the long QT syndrome; HASH2, a transcription factor; and one paternally expressed gene, the insulin-like growth factor 2 (IGF2).128 132–136
The imbalance between imprinted genes (increased IGF2 and diminished H19 and P57KIP2) gives rise to the increase in proliferation of β-cells, a striking feature of focal adenomatous hyperplasia not observed in the diffuse form.128 H19 seems directly or indirectly to modulate cytoplasmic levels of the product of the IGF2 allele and thus the H19 gene seems to be an antagonist to IGF2 in trans.128 Figure 5 illustrates the genetic aetiology of a focal lesion.
The probability for this somatic chromosomal event occurring in a fetus carrying a heterozygous mutation of ABCC8/KCNJ11 of paternal origin is about 1%.128 Recently it was demonstrated that individual patients with focal CHI may have more than one focal pancreatic lesion due to separate somatic maternal deletion of the 11p15 region137 and that some focal lesions have a duplication of the paternal allele on chromosome 11.129
SUMMARY
CHI is a major cause of hypoglycaemia in the newborn and infancy period. So far mutations in seven different genes have been reported which lead to dysregulated insulin secretion. Recent advances have identified the molecular basis of exercise induced CHI (due to mutations in SLC16A1) and highlighted the intriguing link between transient (or persistent HH) and maturity onset diabetes of the young (due to mutations in HNF4A). Rapid genetic analysis for mutations in ABCC8 and KCNJ11 can be used as a powerful tool for differentiating between focal and diffuse disease in some patients and thus aid in patient management. Further genetic studies are required to understand the molecular basis of CHI in patients where no mutations are found in the genes described so far.
REFERENCES
Footnotes
Competing interests: None declared.