Article Text

Abstract
Leber hereditary optic neuropathy (LHON) and autosomal dominant optic atrophy (DOA) are the two most common inherited optic neuropathies and they result in significant visual morbidity among young adults. Both disorders are the result of mitochondrial dysfunction: LHON from primary mitochondrial DNA (mtDNA) mutations affecting the respiratory chain complexes; and the majority of DOA families have mutations in the OPA1 gene, which codes for an inner mitochondrial membrane protein critical for mtDNA maintenance and oxidative phosphorylation. Additional genetic and environmental factors modulate the penetrance of LHON, and the same is likely to be the case for DOA which has a markedly variable clinical phenotype. The selective vulnerability of retinal ganglion cells (RGCs) is a key pathological feature and understanding the fundamental mechanisms that underlie RGC loss in these disorders is a prerequisite for the development of effective therapeutic strategies which are currently limited.
Statistics from Altmetric.com
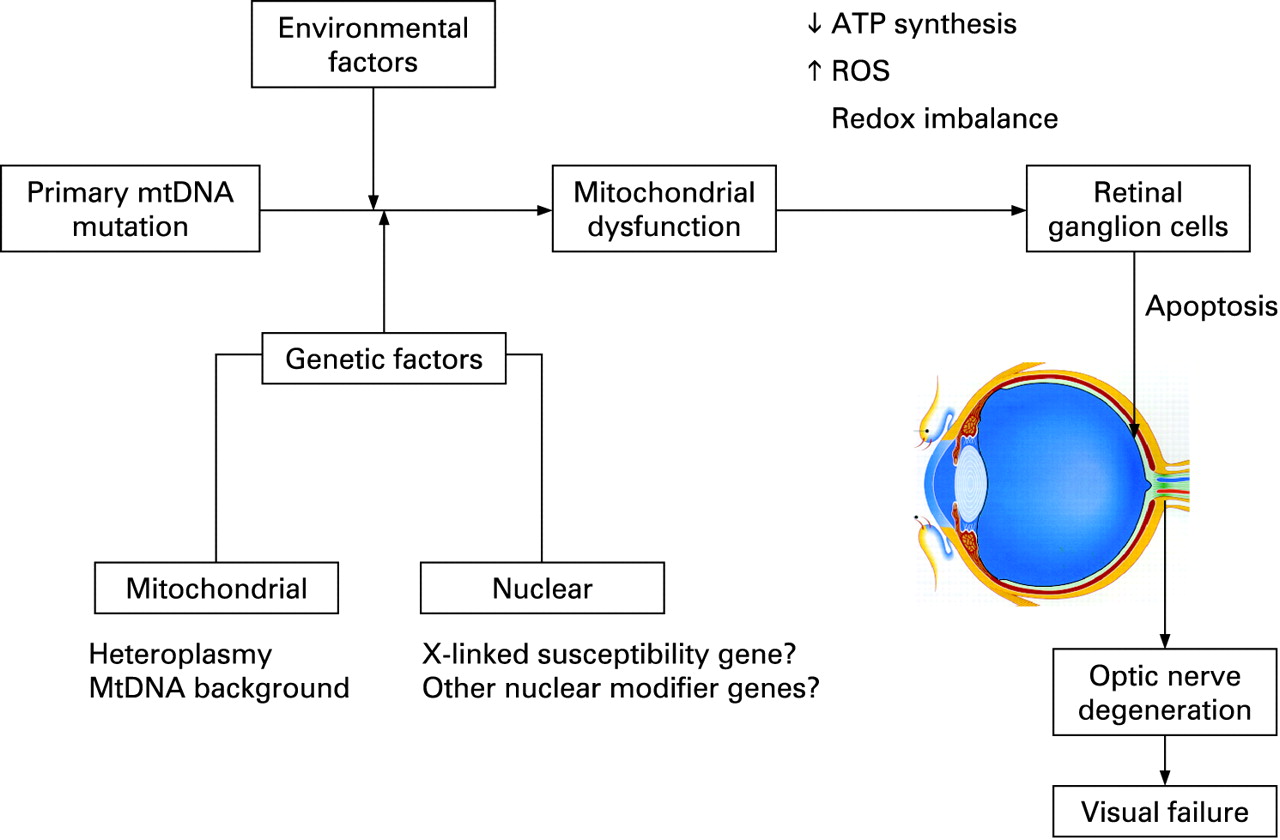
Mitochondrial disorders are a major cause of chronic human disease with an estimated prevalence of 1 in 10 000 in the UK and a further 1 in 200 individuals being at-risk mutational carriers.1 2 Ocular involvement is a prominent feature in this group and often points towards the underlying mitochondrial aetiology, which allows for a more targeted diagnostic approach. Optic nerve dysfunction can be the presenting and only ophthalmological manifestation causing the two most common inherited optic neuropathies encountered in clinical practice, Leber hereditary optic neuropathy (LHON) and autosomal dominant optic atrophy (DOA), which are the focus of this review. In the majority of cases, the pathology in LHON and DOA is limited to a highly specialised group of cells within the eye, the retinal ganglion cells (RGCs), but the phenotype associated with these two conditions is expanding, providing important insights into possible disease pathways leading to optic nerve degeneration and visual failure.
LEBER HEREDITARY OPTIC NEUROPATHY
LHON mutations
LHON (OMIM 535000) was first described as a distinctive clinical entity in 1871 by the German ophthalmologist Theodore Leber (1840–1917).3 He reported a characteristic pattern of visual loss among members of four families and his observations were subsequently confirmed in pedigrees from different populations.4–6 These early studies highlighted several of the salient features of LHON including the maternal transmission of the disease, the predilection of males to lose vision, and the almost exclusive involvement of the optic nerve. The non-Mendelian pattern of inheritance was only fully explained in 1988 when LHON became the first human disease proven to be caused by a point mutation (m.11778G>A) within the mitochondrial genome.7 Over 95% of LHON pedigrees are now known to harbour one of three mitochondrial DNA (mtDNA) point mutations: m.3460G>A, m.11778G>A and m.14484T>C, which all involve genes encoding complex I subunits of the mitochondrial respiratory chain.8 In a meta-analysis of 159 pedigrees from Northern Europe and Australia, m.11778G>A was the most prevalent mutation but there is considerable variation in the relative frequency of these three primary LHON mutations worldwide (table 1). The predominance of m.11778G>A is even more pronounced in the Far East where it accounts for ∼90% of all cases,9 10 and although m.14484T>C is relatively rare, it is the most common mutation found among French Canadians (87%) as a result of a founder event.11 12 Primary mutations have not been identified in a small minority of clinically diagnosed LHON patients, the most likely explanation being that rare pathogenic mtDNA variants are segregating in these families.13 Disease causing mutations have been identified in a proportion of these cases, while other putative LHON mutations require further confirmation as they have only been found in singletons or a single family (table 1).
Epidemiology
LHON is the most common of the primary mtDNA diseases, with a minimum prevalence of 1 in 31 000 affected individuals in the North East of England and 1 in 8500 carriers being at-risk of visual loss.14 Fairly similar figures have been reported in other Caucasian populations, with an LHON prevalence of 1 in 39 000 in the Netherlands and 1 in 50 000 in Finland.15 16 About 2% of visually impaired people on the blind register in Australia are also reported to suffer from LHON.17 The peak age of onset in LHON is between the age of 15–30 years and 95% of carriers who will experience visual failure will do so before the age of 50 years (table 2). However, visual deterioration can occur anytime during the first to the seventh decade of life and LHON should be part of the differential diagnosis for all cases of bilateral, simultaneous or sequential optic neuropathy, irrespective of age and especially in male patients.18 19 Except for one report which found a slight increase in the age of onset in females carrying the m.11778G>A mutation,20 it is generally accepted that neither gender nor mutational status significantly influences the timing and severity of the initial visual loss.11 21–23 Affected individuals are often aware of other affected family members, but up to 40% have no family history. These most likely represent cases where family history is difficult to trace back, given that de novo mutations are rare in LHON.14 24
Clinical features
Pre-symptomatic phase
Fundal abnormalities such as telangiectatic vessels around the optic discs and variable degrees of retinal nerve fibre layer oedema have been documented in some asymptomatic carriers, and these can fluctuate with time. Using optical coherence tomography imaging, thickening of the temporal retinal nerve fibre layer was found in a proportion of unaffected LHON carriers, which provides further evidence that the papillomacular bundle is particularly vulnerable in this disorder.25 26 On more detailed psychophysical testing, some individuals also exhibited subtle impairment of optic nerve function including loss of colour vision affecting mostly the red–green system, reduced contrast sensitivity, and subnormal visual electrophysiological parameters.27
Acute phase
LHON carriers remain asymptomatic until they experience blurring or clouding of vision in one eye. In the vast majority of cases, visual dysfunction is bilateral, the fellow eye becoming affected either simultaneously (25%) or sequentially (75%), with a median inter-eye delay of 6–8 weeks.20 Rare cases of unilateral optic neuropathy in LHON have been reported, with the fellow eye remaining unaffected over a follow-up period of up to 16 years.28 29 Visual acuity reaches a nadir 4–6 weeks after disease onset and it is severely reduced to 6/60 or less. The characteristic field defect is a steep-sided central or centrocaecal scotoma and this can be formally documented using Goldmann or kinetic perimetry. Other clinical features include the early impairment of colour perception but, importantly, pupillary reflexes are preserved and patients usually report no pain on eye movement. Ocular examination during the acute stage provides other diagnostic clues and in classical cases the following abnormalities can be observed: vascular tortuosity of the central retinal vessels, swelling of the retinal nerve fibre layer, and a circumpapillary telangiectatic microangiopathy (fig 1). However, it must be stressed that in ∼20% of LHON cases, the optic disc looks entirely normal in the acute phase.30 31

Chronic phase
The retinal nerve fibre layer gradually degenerates and after 6 months, optic atrophy is a universal feature. If a patient is only seen at this stage, it can be difficult to exclude other compressive, infiltrative and inflammatory causes of a bilateral optic neuropathy, especially if there is no clear maternal family history. In these cases, neuroimaging of the anterior visual pathways is mandatory while awaiting the results of molecular genetic testing.
Visual recovery
Visual recovery is observed in some patients even several years following disease onset. but the chances of improvement are influenced by the patient’s mutational status, being least with the m.11778G>A mutation, highest with the m.14484T>C mutation, and the m.3460G>A mutation having an intermediate visual prognosis (table 2). The recovery in visual parameters is not only restricted to visual acuity, but can also include the development of small islands of normal field (fenestrations) within the central scotoma or a reversal of dyschromatopsia.28 32 33 Positive prognostic factors for visual improvement are an early age of onset (<20 years), subacute presentation with slow progression of the visual deficits, and large optic nerve head surface area.28 34 However, LHON is a devastating disorder with the majority of patients showing no functional improvement and remaining within the legal requirement for blind registration.
Associated features
Although visual failure is the defining feature in this mitochondrial disorder, cardiac arrhythmias and neurological abnormalities such as postural tremor, peripheral neuropathy, non-specific myopathy and movement disorders have been reported to be more common in LHON compared to controls.35–39 These are rarely clinically significant but a small number of LHON pedigrees do have severe neurological deficits (spastic dystonia, ataxia and juvenile onset encephalopathy) in addition to the optic neuropathy. These “LHON plus” syndromes have been linked to various mtDNA mutations in isolated pedigrees from Holland, Australia and North America: A11696G and/or T14596A,40 T4160C,41 and G14459A,42–44 respectively. Two mtDNA complex I mutations point mutations, m.3376G>A45 and m.3697G>A,46 have also recently been identified in individuals with overlap clinical features of both LHON and MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes). Interestingly, a significant minority of Caucasian LHON carriers, predominantly females with the m.11778G>A mutation, develop clinical and neuroimaging features indistinguishable from multiple sclerosis (MS), including unmatched oligoclonal bands in the cerebrospinal fluid (Harding’s disease).47–50 It is currently not known whether the prevalence of this MS-like illness in LHON is higher than expected due to the chance occurrence of these two disorders, and although controversial, some investigators have argued for a potential role of autoimmunity in the pathophysiology of this mitochondrial disorder.51–55
Diagnosis
A tentative diagnosis of LHON can usually be made on clinical grounds, especially if classical ophthalmological features are present and a clear maternal history is elicited. Molecular genetic testing on a blood DNA sample, however, remains the gold standard and will confirm that the patient harbours one of the three primary mtDNA LHON mutations, with implications for future genetic counselling. If indicated, electrophysiological studies, including pattern electroretinograms (PERGs) and visual evoked potentials (VEPs), can be carried out to exclude retinal pathology and confirm optic nerve dysfunction.56 An electrocardiogram is also recommended to exclude a pre-excitation syndrome which has been documented in LHON, although such a finding is rare and does not require any intervention in the absence of cardiac symptoms.30 31 Computed tomography (CT) and magnetic resonance imaging (MRI) scans are usually normal in LHON, but there are reports of non-enhancing high signals within the optic nerve and sheath distension, secondary to slight oedema or gliosis in the atrophic phase.57–62
Biochemical features
Oxidative phosphorylation (OXPHOS) provides for most of the cell’s adenosine triphosphate (ATP) requirements and this is achieved by a chain of five respiratory complexes situated on the inner mitochondrial membrane. Since all three primary LHON mutations involve complex I subunits, one would expect respiratory chain function to be compromised, leading to a deficit in ATP synthesis and RGC degeneration as a consequence of energy failure. However, both in vitro and in vivo biochemical studies have produced conflicting results regarding the extent of respiratory chain dysfunction in LHON (table 3). In a small number of in vivo studies using phosphorus magnetic resonance spectroscopy (31P-MRS), the most consistent defect of mitochondrial function was identified in persons with the m.11778G>A mutation and none among those with the m.3460G>A mutation.63–67 A striking conclusion from all these biochemical studies is that no significant difference between affected and unaffected individuals with a disease causing LHON mutation could be demonstrated. But as none of these studies have been performed directly on RGCs and the causative biochemical mechanisms could be highly tissue-specific, further studies are warranted.
Neuropathology
These functional studies also raise important issues regarding the cell specific ocular pathology in LHON which is limited to the RGC layer, with sparing of the retinal pigment epithelium and photoreceptors. There is pronounced cell body and axonal degeneration, with associated demyelination and atrophy observed from the optic nerves to the lateral geniculate bodies. Experimental data indicate impaired glutamate transport,68 oxidative stress69 70 and increased mitochondrial reactive oxygen species (ROS)71 within RGCs and support an apoptotic mechanism of cell death.72 73 LHON patients also have reduced α-tocopherol/lipid ratios and high levels of 8-hydroxy-2-deoxygaunosine in blood leucocytes, both biological markers of increased free radical production.74 75 However, the selective vulnerability of RGCs in LHON still remains unexplained, and this area of research has been greatly hampered by the lack of access to diseased human tissues, the retina and optic nerve not being amenable to biopsies.
Animal models
The development of faithful animal models in LHON is therefore critical but there is still no murine model where the primary LHON mutations have been successfully introduced within the mitochondrial genome. In spite of these technical challenges, significant advances have been made over the past decade and there are currently three experimental paradigms, all of which disrupt OXPHOS and recapitulate the optic nerve degeneration seen in LHON: (1) intravitreal injection of a respiratory chain poison such as rotenone76; (2) downregulation of nuclear encoded complex I subunits (for example, NFUFA1) with specific mRNA-degrading ribozymes77; and (3) allotropic expression of mutant subunits (for example, MTND4) which are then imported into the mitochondria.78
Incomplete penetrance
An intriguing feature of LHON is that only ∼50% of males and ∼10% of females who harbour one of the three primary mutations actually develop the optic neuropathy. This incomplete penetrance and predilection for males to lose vision imply that additional genetic and/or environmental factors must modulate the phenotypic expression of LHON (fig 2). Alternatively, the gender bias could also result from a combination of subtle anatomical, hormonal and physiological variations between males and females.

Mitochondrial genetic factors
Heteroplasmy
Depending on their metabolic demands, cells can contain anywhere between 100–10 000 mitochondria, and with 2–10 mtDNA molecules in each mitochondrion, this results in a very high copy number per cell. In most LHON pedigrees, the primary mutation is homoplasmic—that is, every mtDNA molecule harbours the mutant allele. By contrast, 10–15% of LHON carriers are thought to be heteroplasmic, with one mtDNA sub-population carrying the wild type allele.14 20 79 Although limited and retrospective, the available data suggest that heteroplasmy contributes to incomplete penetrance, with the risk of blindness being minimal if the mutational load is <60%.80 However, quantifying the level of heteroplasmy for the purpose of pre-symptomatic testing is limited as the majority of individuals with a LHON mutation are homoplasmic.
MtDNA haplogroups
MtDNA accumulates mutations ∼10 times faster than nuclear genome, resulting in a high degree of polymorphism.81 Because human mtDNA is strictly maternally inherited and does not recombine, polymorphisms have accumulated sequentially along radiating female lineages as women migrated out of Africa into the different continents ∼150 000 years ago.82 Reflecting its evolution, a number of stable polymorphic variants cluster together in specific combinations referred to as haplogroups, with individuals of European ancestry belonging to one of nine haplogroups: H, I, J, K, T, U, V, W and X.83 84 A recent meta-analysis of 159 European LHON pedigrees indicated that the risk of visual loss for the three primary LHON mutations is influenced by the mtDNA background.85 The risk of visual failure was greater when the m.11778G>A and m.14484T>C mutations arose on haplogroup J, whereas individuals with the m.3460G>A mutation were more likely to experience visual loss if they belonged to haplogroup K. On the other hand, individuals with the m.11778G>A mutation had a lower risk of visual loss when the mutation arose on haplogroup H. Haplogroups H, J and K are all defined by non-synonymous, polymorphic substitutions in the MT-CYB gene which codes for cytochrome b, the only mitochondrially encoded subunit of complex III. Recent experimental data support the existence of stable respiratory chain supercomplexes, one of which consists of a complex I monomer physically interacting with a complex III dimer. Although speculative, the haplogroup associated amino acid substitutions within cytochrome b could therefore influence the risk of visual failure by modulating the biochemical consequences of the primary LHON mutations through an effect on the stability of these putative I-III supercomplexes.85–87 In support of this hypothesis, cybrid cell lines carrying the m.11778AG>A mutation on a haplogroup J background had a lower oxygen consumption and a longer doubling time compared to non-haplogroup J cell lines.88 However, haplogroup J was not found to further impair mitochondrial OXPHOS in the brain and skeletal muscle of patients harbouring the m.11778G>A mutation with 31P-MRS measurements,66 and a study of South-East Asian LHON pedigrees found no association between specific mtDNA haplogroups and the risk of visual loss.89 These contradictory findings reflect the need for additional studies to clarify the significance of the mtDNA background on LHON penetrance.
Nuclear genetic factors
The predominance of affected males in LHON cannot be explained by mitochondrial inheritance and segregation analysis suggests the existence of a recessive X-linked susceptibility gene acting in synergy with the mtDNA mutation to precipitate the optic neuropathy.90–92 In the Bu and Rotter model, the development of blindness in males is consistent with the simultaneous inheritance of an X-linked visual loss allele and the primary LHON mutation, whereas females are affected either if they are homozygous at the susceptibility locus (40%) or heterozygous with skewed X chromosome inactivation of the wild-type allele (60%). Several studies have, however, failed to demonstrate any skewed X chromosome inactivation in affected female carriers, albeit in blood leucocytes and not in RGCs which are the affected tissues in LHON.93–95 Initial attempts to identify this X-linked susceptibility locus by standard linkage analysis were unsuccessful,96–99 but two recent studies using a larger number of more extensively defined LHON pedigrees found two overlapping disease loci with highly significant LOD scores at Xp21–Xq21100 and Xq25–27.2.101 Although the actual causative gene in this region of interest has not yet been identified, a high risk haplotype [DXS8090(166)-DXS1068(268)] at Xp21 was defined which increased the risk of visual failure ∼35-fold for the m.11778G>A and m14484T>C mutations but not for m.3460G>A.100 The possibility of other autosomal nuclear modifier genes in LHON has not been excluded and the genetic aetiology of LHON might prove even more complex, with epistatic interaction of these multiple nuclear susceptibility loci and genetic heterogeneity.
Environmental factors
Five pairs of monozygotic twins harbouring a primary LHON mutation have been reported in the literature, and in two cases the twins have remained discordant.20 21 24 102–104 Although there is always the possibility that the unaffected sibling will lose vision later on in life, the existence of discordant monozygotic twins strongly suggests that environmental factors also contribute to penetrance. There are several reports of an increased risk of visual loss among LHON carriers with high tobacco and alcohol consumption,105–108 but the largest case–control study to date has failed to confirm this association.109 There are also anecdotal reports of nutritional deprivation, exposure to industrial toxins, antiretroviral drugs, psychological stress or acute illness precipitating the onset of blindness in LHON.108 110–112 Of note, in some pedigrees the penetrance of LHON seems to be decreasing, falling to 1% and 9% in younger generations of two large, multi-generational pedigrees from Australia113 and Brazil,108 114 respectively. Both carry homoplasmic levels of the m.11778G>A mutation and this phenomenon has been ascribed to improved environmental and socio-economic factors. However, a much larger epidemiological study of 3613 LHON carriers from multi-generational pedigrees failed to detect a change in the penetrance of the three primary LHON mutations. The role of environmental triggers in LHON remains largely unanswered and more robust epidemiological data are needed, which will necessitate a multicentre collaborative effort in order to collect sufficient number of subjects for analysis.
Treatment
No generally accepted measures have been shown to either prevent or delay the onset of blindness in LHON, but for general health reasons LHON carriers should be advised to moderate their alcohol intake and stop smoking. In two small case series, oral administration of idebenone, a synthetic analogue of coenzyme Q10, and vitamin B12 and C supplementation led to faster and greater visual recovery among affected individuals.115 116 However, a recent study has not found any improved visual prognosis from idebenone and multivitamin supplementation, and properly conducted treatment trials are needed before such a regimen can be advocated.117 The use of brimonidine eye drops, which is thought to have anti-apoptotic properties, was also unsuccessful in preventing second eye involvement in recently affected patients with unilateral optic neuropathy.118 The long term management of visually impaired patients remains supportive, with provision of visual aids and registration with the relevant social services.
Genetic counselling
It is important to stress to LHON carriers that it is not possible to predict accurately whether or when they will become affected. Despite these caveats, the two main predictive factors for visual failure remain age and gender, with males having about a 50% lifetime risk of blindness compared to only 10% for females, and these approximate figures can be further refined based upon the patient’s age. From published age dependent penetrance data, most patients experience visual loss in their late teens and 20s and the probability of becoming affected decreases with increasing age, being minimal once past the age of 50 years (table 2). Once a primary LHON mutation has been identified in a proband, other maternally related family members can be offered molecular genetic testing to exclude the possibility of a de novo mutation, which is rare. Since LHON shows strict maternal inheritance, male carriers can be reassured that none of their children will inherit the mtDNA mutation whereas female carriers will transmit the pathogenic mutation to all of their offspring. Since most mothers are homoplasmic, their children will only harbour the mutant species, but the situation is more complex for a heteroplasmic mother as she could transmit a higher or a lower level of the mutation to a particular offspring, which will impact on the latter’s risk of visual failure. Although the mutant level can be determined and there is evidence that a mutational threshold of ∼60% in blood is necessary for disease expression, genetic counselling for these unaffected heteroplasmic carriers remains difficult. For similar reasons, the prenatal genetic testing of heteroplasmic women with amniocentesis or chorionic villus sampling (CVS) would be difficult to interpret.
DOMINANT OPTIC ATROPHY
Clinical features
The clinical features of DOA (OMIM 165500) were first described in one British family by Batten in 1896119 120; the phenotype was further clarified by Kjer in his extensive study of Dutch families in the 1950s,119 120 distinguishing it from LHON with which the disease was often confused. The prevalence of DOA is not well established and robust estimates based on molecular confirmation are not available, although a historical figure of 1 in 50 000 among Caucasians is often quoted in the literature.121 It is thought to be the most common inherited optic neuropathy in the Netherlands, with a population frequency of 1 in 12 000, and this much higher prevalence has been linked to a mutational founder event.122
The onset of symptoms in DOA is relatively insidious. In pre-molecular case series, 13–25% of patients with optic atrophy were visually asymptomatic and were only identified through contact tracing via other affected family members.123 124 Classically, the visual decline starts in the first two decades of life, but there is a pronounced inter- and intra-familial variability in the severity of visual symptoms, which makes genetic counselling difficult. Visual acuity can range from 6/6 to the detection of hand movement only, and the rate of progression of visual loss is not easy to predict, with 19–50% of patients experiencing further, albeit slow, deterioration on long term follow up.125–129 Although the overall visual prognosis is better when compared to LHON, with a mean visual acuity of 6/24–6/36, DOA results in significant visual impairment with about half of all affected individuals failing the driving standards and 13–46% registered as legally blind.130–132
The predominant colour defect in DOA is a generalised dyschromatopsia, involving both the blue–yellow and red–green axes, with a minority of patients having pure tritanopia (<10%), which was once considered to be a pathognomonic feature of DOA.133 Central, centrocaecal and paracentral scotomas are the most common field abnormalities with sparing of the periphery, findings consistent with the primary involvement of the papillomacular bundle in this condition. Interestingly, as in LHON, there is usually no afferent pupillary defect, suggesting that the retino-tectal fibres sub-serving the pupillary light reflex are less susceptible to the downstream effects of both the LHON mtDNA mutations and the causative nuclear genetic defects in DOA.134 However, both magnocellular and parvocellular RGC pathways seem to be similarly affected, although this requires further investigation.127 131
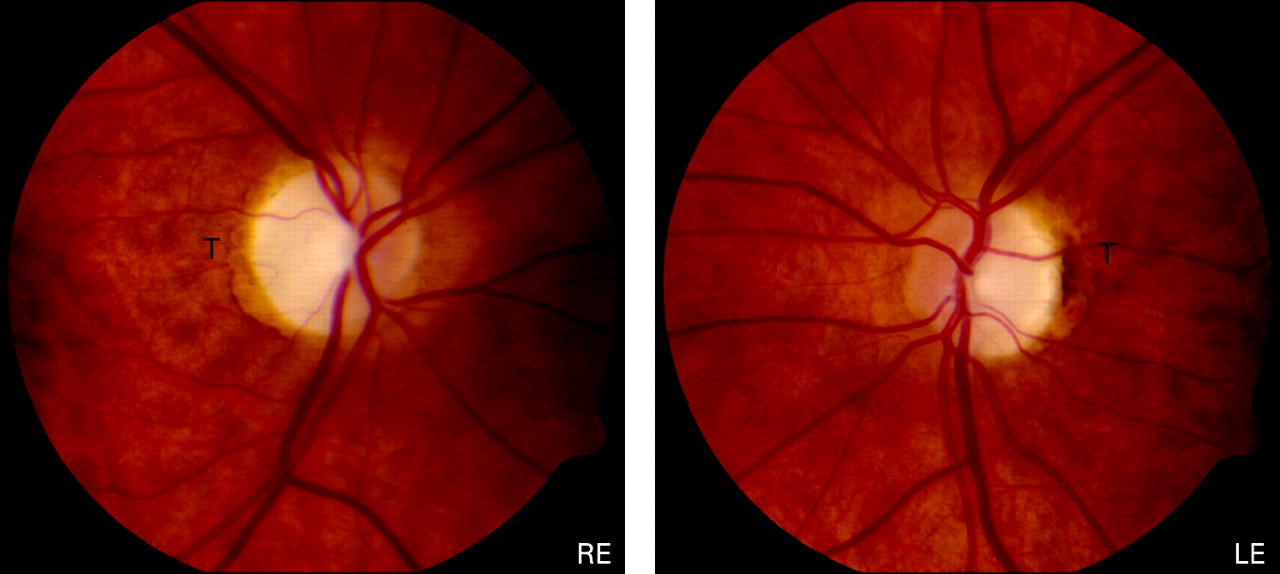
The optic disc pallor in DOA falls into two main categories: diffuse pallor involving the entire neuro-retinal rim in about half of all cases, and a temporal wedge in the remainder (fig 3).123 135 However, disc pallor can be subtle and 29% of affected patients had normal looking optic discs in one case series, highlighting the need to look carefully for other features of optic nerve dysfunction when assessing patients with a possible diagnosis of DOA.132 Other common optic disc findings include saucerisation (79%), peripapillary atrophy (69%) and a cup to disc ratio >0.5 (48%).131 135 136 The measurement of circumpapillary retinal nerve fibre layer thickness using optical coherence tomography (OCT) could also prove a useful adjunct in the diagnostic work-up of DOA, with recent studies showing a typical profile with bilateral symmetrical thinning around the optic disc, most pronounced in the temporal quadrant.137 138

{kind=link}
{kind=link}
{kind=link}
Ocular pathology
Postmortem studies of two patients with DOA identified similar histopathological changes, with diffuse atrophy of the RGC layer, loss of myelin and fibrillary gliosis along the anterior visual pathways extending to the lateral geniculate body.139 140 More recent MRI data from patients with DOA have also confirmed significant tissue loss and thinning of the optic nerve along its entire length.141 Although less pronounced, the underlying ocular pathology in DOA is therefore remarkably similar to LHON, with the primary loss of RGCs leading to ascending optic atrophy.
Visual electrophysiological findings are well documented in DOA and provide additional evidence for the primary loss of RGCs and the sparing of the outer retinal layers.133 142 143 It can therefore be a useful ancillary test when determining affected status in borderline DOA cases and also in excluding a primary retinal process such as early cone dystrophy. VEPs are either absent or, if traces are recordable, they are of low amplitudes with abnormal latencies. PERGs can be within the normal range in up to 40% of clinically affected individuals but usually demonstrate an abnormal P50:N95 ratio, with selective depression of the N95 negative wave amplitude confirming a primary optic nerve pathology. Additional involvement of the P50 component correlates with the severity of visual loss, but PERGs are not extinguished even in cases where visual acuity is reduced to detection of hand movements or worse.
Molecular genetics
The majority of DOA families show linkage to the OPA1 locus at 3q28–q29, and in 2000 two independent research groups identified pathogenic mutations in the OPA1 gene.144 145 The proportion of OPA1 positive families is ∼60% (range 32–89%), the lower detection rates in some of these case series reflecting the inclusion of singleton cases, a heterogeneous group that is more likely to include non-inherited forms of optic neuropathy, and the use of less sensitive mutation screening protocols such as single strand conformational polymorphism (SSCP) analysis.146 147 Interestingly, a recent report suggested that large scale rearrangements of entire OPA1 coding regions could account for up to 20% of all OPA1 negative cases.148
The causative nuclear defects in the remaining families with DOA have not yet been identified, but a small number of families have been mapped to other chromosomal loci—OPA3, OPA4, OPA5 and OPA7, of which only the OPA3 gene has been characterised (table 4). The OPA3 gene was originally identified in eight Iraqi Jewish families with an autosomal recessive form of optic atrophy, associated with neurocognitive deficits, elevated urinary excretion of 3-methyl glutaconic acid, and increased plasma 3-methylglutaric acid levels (type III 3-methylglutaconic aciduria or Costeff syndrome).149–151 However, pathogenic mutations in the OPA3 gene have also been identified in two French families segregating both DOA and premature cataract in an autosomal dominant mode of inheritance (ADOAC).152 153 The Opa3 protein is located in the mitochondrial inner membrane but its exact function remains to be clarified. Preliminary findings in cultured fibroblasts from a patient with ADOAC indicate an increased susceptibility to apoptosis, and one can speculate that a similar mechanism is leading to RGC dysfunction via disruption of the mitochondrial respiratory chain.152 154 155
OPA1 mutations
The OPA1 gene consists of 30 exons spanning over 100 Kb of genomic DNA and it codes for a 960 amino acid, dynamin related GTPase protein located within the inner mitochondrial membrane. Alternative splicing of exons 4, 4b and 5b result in eight different mRNA isoforms, and both their functional relevance and subcellular localisation are currently being investigated.156 Over 140 pathogenic mutations have been identified and these cluster in two specific regions: the GTPase region (exons 8–15) and the C-terminus which is the proposed site of the GTPase effector domain. The majority of OPA1 mutations (∼50%) lead to premature termination codons (PTCs) as a result of nonsense mutations or frameshifts from small insertions, deletions or splice site mutations (eOPA1 database at http://lbbma.univ-angers.fr/lbbma.php?id = 9).157 These truncated mRNAs are unstable and get degraded by specific pathways (nonsense mediated mRNA decay), which are in-built protective cellular mechanisms against mutant proteins with possible dominant negative effects.158–160 The reduced Opa1 protein expression levels observed in these cases support the role of haploinsufficiency in DOA and this is further substantiated by one family with a microdeletion resulting in complete loss of one copy of the OPA1 gene.161 However, ∼30% of OPA1 mutations are missense mutations within or close to the GTPase domain and these could exert their pathogenic effect via a deleterious, gain of function mechanism.162–164
Gene expression
The spatial localisation and expression pattern of the Opa1 protein have been examined in a wide range of post-mitotic human and murine tissues. The Opa1 protein is highly expressed in the RGC layer but it is also found at comparable levels in the photoreceptor, inner and outer plexiform retinal layers.165 166 In the human optic nerve, Opa1 was detected along the axonal tracts both in the pre- and post-lamina cribosa regions.167 168 The Opa1 protein is ubiquitous and abundant levels have been identified in non-ocular tissues such as the inner ear and various areas of the human brain, with a similar distribution pattern of the different isoforms.169 170 Overall, these immunohistochemical studies indicate that differential tissue expression of the OPA1 gene or its isoforms do not seem to underlie the selective vulnerability of RGCs in DOA.
Protein function
The Opa1 protein is part of the large, dynamin GTPase family of mechanoenzymes and it was first identified in a screen for nuclear genes required for mtDNA maintenance in the budding yeast Saccharomyces cerevisiae. Both the human and yeast (Mgm1+) homologues show a high degree of evolutionary conservation and functional studies in DOA have revealed several other important cellular roles in addition to mtDNA maintenance.171 172
Mitochondrial maintenance
Opa1 is an important pro-fusion protein and works in tandem with other members of the dynamin related mitofusin family (mfn-1 and mfn-2) to balance the pro-fission effects of other GTPases such as Drp1 and Fis-1.173 174 It is therefore not surprising that mitochondrial network disruption is a key pathological feature seen in fibroblasts from DOA patients and other tissue cultures, including RGCs, where the expression of the Opa1 protein has been disrupted—for example, by small interfering RNAs.162 170 175 176 Instead of a typical elongated, filamentous mitochondrial network, the latter becomes highly fragmented, with isolated mitochondria showing aberrant balloon-like enlargements. Transmission electron microscopy (TEM) also confirms altered mitochondrial ultrastructure with abnormal mitochondrial cristae organisation and paracrystalline inclusion bodies.162
Fusion is postulated to subserve a protective biological function by allowing the exchange and complementation of mitochondrial contents.177 178 In this respect, neuronal cells with deficient mitochondrial fusion show a loss of mtDNA nucleoids and this important finding provides a possible disease mechanism, with the reduced expression of essential, mtDNA encoded, respiratory chain subunits resulting in a bioenergetic deficit, increased ROS levels and a greater susceptibility to undergo apoptosis.179 180 These deleterious consequences could also contribute to the formation and clonal expansion of secondary mtDNA abnormalities such as mtDNA deletions, which have recently been identified in a subgroup of DOA families with a more complex multi-system involvement in addition to the optic neuropathy.162–164
Oxidative phosphorylation
Impaired mitochondrial biogenesis is central to the pathophysiology in DOA and there is good experimental evidence to support a predominant complex I defect. There is reduced mitochondrial membrane potential and ATP synthesis in fibroblast cultures carrying pathogenic OPA1 mutations,181 182 and in vivo disturbance of oxidative metabolism was evident in the calf muscle of patients with DOA using 31P-MRS.183 Immunoprecipitation studies also suggest that the Opa1 protein, in conjunction with other structural proteins such as the apoptosis inducing factor (AIF), interacts directly with complexes I, II and III and plays an important role in the assembly and stabilisation of their various component subunits.176 This provides another causal link between OPA1 mutations and the resulting mitochondrial respiratory chain defect in DOA.
Apoptosis
Apoptosis is the final common pathway leading to RGC loss in DOA and cell death is likely be complex, being triggered by a combination of several interacting factors. Opa1 is processed by various, inner membrane proteases which include the presenilin associated rhomboid-like protein (Parl) and paraplegin, and this proteolytic cleavage results in a soluble, intermembrane form in addition to the integral, membrane bound form.184–186 These two proteins combine into oligomers which modulate the morphology of the inner mitochondrial membrane and the tightness of the cristae junctions, a process independent of the role of Opa1 in controlling fusion.187 Downregulation of Opa1 leads to aberrant cristae remodelling and the release of cytochrome c which is normally sequestered in the narrow junctions within the cristae.175 188 This will either be sufficient on its own to induce the apoptotic cascade or will sensitise the cell to other pro-apoptotic stimuli such as AIF, increased ROS or the dissipation of the mitochondrial membrane potential.
Animal models
There are now two established mouse models of DOA, with heterozygous mutations in exon 8 (c.1051C>T) and intron 10 (c.1065+5g>a) of the OPA1 gene.189 190 These two mutations are truncative, resulting in a 50% reduction in the expression of the Opa1 protein, and therefore represent a haploinsufficiency disease mechanism. In both models, homozygous mutant mice (OPA1−/−) died in utero during embryogenesis, highlighting the central role played by the Opa1 protein in early development. Heterozygous OPA1+/− mice faithfully replicated the human phenotype exhibiting a slowly progressive optic neuropathy and demonstrating objective reduction in visual function on psychophysical testing. There was a gradual loss of RGCs, leading to thinning of the retinal nerve fibre layer, and the surviving optic nerve axons had an abnormal morphology with swelling, distorted shapes, irregular areas of demyelination and myelin aggregates. Mitochondria within these axons showed disorganised cristae structures on TEM and cultured fibroblasts showed fragmentation of the mitochondrial network. These two OPA1 mouse models represent powerful tools for dissecting the pathways mediating the preferential loss of RGCs in DOA, by allowing functional studies to be performed directly on these specialised cells, something which is not possible in humans given the lack of ocular tissues. These mutant mice will also prove useful when investigating the potential therapeutic benefit of future biological agents which could be injected into the vitreous cavity, allowing direct access with the RGC layer.
Expanding phenotype
The hallmark of DOA is bilateral visual failure, but sensorineural deafness is a well reported association which is more commonly observed with some pathogenic mutations such as the p.R445H mutation.191–193 In his original description, Kjer also documented neurodevelopmental abnormalities in 10% of his Dutch cohort, although this has not been reported in other populations.120 125 More recently, DOA families have been described where the optic atrophy was segregating with additional ocular and extraocular features such as progressive external ophthalmoplegia, ptosis, myopathy, ataxia, neuropathy, and an MS-like disorder.162–164 194 These syndromal variants of DOA, so-called “DOA plus”, have been linked with the accumulation of multiple mtDNA deletions, a finding consistent with the presence of cytochrome c oxidase (COX) deficient fibres in limb muscle biopsies from affected individuals.195 All of the causative OPA1 mutations in these families were missense mutations with most, but not all of them, within the catalytic GTPase site of the protein. Although the actual proportion of families with these “DOA plus” phenotypes is as yet unknown, clinicians need to be aware of these additional clinical features as these can be subtle and therefore easily missed if not looked for specifically.
Genetic counselling
There is currently no treatment to influence the disease process in DOA and clinical management, as for LHON, is supportive. Despite DOA being an autosomal dominant Mendelian disorder, genetic counselling for mutational carriers is difficult because of the pronounced inter- and intra-familial variability in the visual phenotype. There are no definite genotype–phenotype correlations but missense mutations within the GTPase protein domain are more likely to result in a complex, multi-systemic involvement, although it must be stressed that this observation requires further investigation in a larger cohort of DOA families.
With the availability of molecular testing for OPA1 becoming more accessible, an increasing number of individuals with pathogenic mutations are being identified who are otherwise visually unaffected. The penetrance is >80% in well characterised, multi-generational families but figures as low as 43% have been reported, probably reflecting the different assessment criteria used (range 43–100%).132 196 197 This incomplete penetrance together with the variable clinical expressivity in both pure DOA and “DOA plus” families clearly imply that other, as yet unidentified, secondary factors are potentiating the deleterious effects of the OPA1 mutations.
MITOCHONDRIAL OPTIC NEUROPATHIES
The concept of inherited mitochondrial optic neuropathies is expanding with evidence of impaired mitochondrial function in other genetic diseases where optic nerve dysfunction is a recognised clinical feature (table 5). These include: (1) Friedreich’s ataxia where up to a third of cases have an optic neuropathy198 199; (2) hereditary motor and sensory neuropathy type 6 (HMSN-6), a variant of Charcot–Marie–Tooth (CMT) disease defined by the presence of both optic atrophy and peripheral neuropathy200 201; and (3) the hereditary spastic paraplegias (HSP).202–204
Glaucoma is the second most common cause of blindness in developed countries and accounts for about 10% of all blind registration in the UK.205 It is a primary, acquired optic neuropathy with a strong genetic component and OPA1 mutations have been identified in a number of patients initially diagnosed with normal tension glaucoma, highlighting the similarities in optic disc features shared with DOA.206 207 It is of note therefore that some studies have shown an association between the risk of developing glaucoma and certain OPA1 polymorphic variants,208–210 with other investigators reporting mtDNA abnormalities in their glaucoma cohorts, such as an increased mtDNA copy number and reduced respiratory chain activities in peripheral blood lymphocytes.211 Although further studies are needed, these findings suggest a possible mitochondrial influence on the pathogenesis of glaucoma.
UNIFYING HYPOTHESIS
The common theme in the various optic neuropathies described in this review is the vulnerability of RGCs to mitochondrial dysfunction. Although there is a high level of mitochondrial enzyme activity in RGCs,212 this phenomenon cannot be explained by a simple energetic deficit since photoreceptors have a much higher oxidative demand than RGCs and other mitochondrial disorders characterised by more severe complex I defects do not universally cause optic atrophy. It is possible that RGCs are preferentially involved because they are more sensitive to subtle imbalances in cellular redox state or increased ROS levels, but an attractive hypothesis implicates the differential mitochondrial concentration observed at the lamina cribosa.213 The lamina cribosa is a perforated collagen plate that marks the anatomical transition from the unmyelinated (pre-laminar) to the myelinated (post-laminar) segment of the human optic nerve. The pre-laminar section has a much higher concentration of mitochondria to support the higher energy demands of unmyelinated nerve conduction and it is likely that active processes involving the cytoskeletal architecture are needed to maintain this sharp mitochondrial gradient.214 215 Pathological mechanisms which disrupt this unique structural feature would lead to impaired axonal transport, as seen in CMT179 216 217 and HSP,218 219 and set up a vicious circle with fragmentation of the mitochondrial network at the lamina cribosa exacerbating even subtle mitochondrial energy deficits and eventually precipitating apoptotic cell death.
CONCLUSION
LHON and DOA show an intriguing degree of clinical and mechanistic overlap, with both disorders caused by the selective degeneration of the RGC layer. They are the two most common inherited optic neuropathies and they provide strong evidence that the maintenance of RGCs is heavily dependent upon normal mitochondrial function. This is further substantiated by recent studies pointing towards a mitochondrial link in sporadic glaucoma and other genetic disorders where optic nerve dysfunction is a prominent clinical feature. Although major advances have been achieved in the two decades since the primary LHON mutations were identified, several key questions remain unanswered. What secondary factors account for the notable incomplete penetrance and male bias in LHON? What explains the variable disease expression in DOA, and why is there no gender bias in this disorder, given the similarity to LHON? What are the causative nuclear genes in OPA1-negative families and will they also involve mitochondrial dysfunction? What mechanisms underpin the preferential loss of RGCs in these mitochondrial optic neuropathies? The characterisation of recently developed animal models and future genetic and functional studies will hopefully reveal important pathophysiological pathways amenable to therapeutic interventions.
Acknowledgments
PFC is a Wellcome Trust Senior Fellow in Clinical Science and PYWM is an MRC Clinical Research Fellow.
REFERENCES
Footnotes
Competing interests: None.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Linked Articles
- Miscellaneous