Article Text

Abstract
Hereditary non-polyposis colorectal cancer (HNPCC) is an autosomal dominant syndrome characterised by a predisposition to early onset colorectal, endometrial and other cancers. The tumours typically exhibit microsatellite instability due to defective mismatch repair. HNPCC is classically caused by heterozygous loss-of-function mutations within the mismatch repair genes MLH1, MSH2, MSH6 and PMS2, but no pathogenic mutations are identified in a third of cases. In recent years, constitutional epimutations of the MLH1 gene, characterised by soma-wide allele specific promoter methylation and transcriptional silencing, have been identified in a handful of mutation negative HNPCC cases. In contrast to genetic mutations, MLH1 epimutations are reversible between generations and thus display non-Mendelian inheritance. This review focuses on the aetiological role of constitutional MLH1 epimutations in the development of HNPCC related cancers. The molecular characteristics, clinical ramifications and potential mechanism underlying this defect are discussed. Recommendations for the selection of cases warranting screening for MLH1 epimutations are proffered.
Statistics from Altmetric.com
Hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome (OMIM 120435) is an autosomal dominant DNA mismatch repair deficiency syndrome characterised primarily by the early development of colorectal and endometrial cancers, with an elevated risk of small bowel, ureter, renal pelvis, biliary tract, brain, gastric and ovarian cancer.1 HNPCC is classically caused by heterozygous deleterious mutations within either of the mismatch repair genes MLH1, MSH2, MSH6 or PMS2, but most frequently within MLH1 and MSH2.2 3 These genes encode the key components of the mismatch repair system, which comprises two main heterodimeric protein complexes: the MutL homologue (MLH1 and PMS2), and the MutS homologue (MSH2 and MSH6).4 Tumourigenesis usually follows Knudson’s “two hit” model in which the predisposing germline mutation (first hit) is followed by somatic loss of function of the remaining normal allele (second hit).5 HNPCC associated tumours almost invariably display microsatellite instability (MSI) as a direct consequence of impaired mismatch repair activity.6 However, pathogenic sequence mutations of the mismatch repair genes fail to be identified in about one third of cases meeting the clinical criteria for HNPCC.7 In recent years, an aetiological role for the epigenetic inactivation of the mismatch repair genes MLH1 and MSH2 has been revealed in rare cases with an HNPCC phenotype but normal sequence of the mismatch repair genes.8 9 10 11 12 13 14 15 16 17 This type of defect was initially termed a “germline epimutation”,10 but is referred to as a constitutional epimutation herein. Constitutional epimutations are characterised by methylation of a single allele of the promoter accompanied by transcriptional silencing of the affected allele in the normal somatic tissues, in an otherwise intact gene.10 11 They appear to confer a similar phenotype as sequence mutations of the same gene, thus can serve as an alternative aetiological mechanism for HNPCC. However, constitutional epimutations of MLH1 are reversible during meiosis and so display non-Mendelian inheritance, in contrast to the strict autosomal dominant inheritance pattern associated with genetic mutations of the mismatch repair genes.12 The mechanism that underlies this defect and its inheritance pattern remain to be elucidated. In the following sections, we discuss the role of constitutional epimutations in HNPCC, with the focus on MLH1. We summarise what is currently known about constitutional MLH1 epimutations from a molecular and clinical perspective. Of clinical importance, we provide recommendations for the triaged selection of cases for molecular screening to identify carriers of this defect and discuss the inheritance pattern associated with it. Finally, we explore the potential mechanisms that might underlie constitutional MLH1 epimutations.
Clinical and genetic diagnosis of HNPCC
For many years, HNPCC cases warranting genetic screening for germline mutations of the mismatch repair genes were identified on the basis of the Amsterdam I criteria, which represented the “gold standard” for a clinical diagnosis of HNPCC (three relatives with colorectal cancer present in at least two successive generations, one of whom must be a first degree relative of the other two; colorectal cancer diagnosed in at least one relative below the age of 50 years).18 19 These criteria have been progressively revised to incorporate additional clinical and molecular characteristics of HNPCC as a fuller picture of the phenotypic spectrum associated with mismatch repair mutations emerged, but each has in common early age of onset and the histological appearance of tumours, with a familial component required in some of the schemes.18 19 20 21 22 The less stringent revised Bethesda guidelines were devised to take into account extracolonic cancers, the lack of a significant family history due to small family size or the occurrence of de novo mutations, and tumour histology.19 20 21 In the latter guidelines, genetic screening for a germline mutation is recommended if the tumour exhibits MSI and the proband meets one or more of the clinical criteria. HNPCC is clinically heterogeneous depending on which of the mismatch repair genes is affected. The phenotype associated with mutations of MLH1 and MSH2 has been described as “classic HNPCC”, with an estimated lifetime risk of developing colorectal cancer of ∼80% in males and ∼50% in females.23 24 25 26 In females the risk of developing endometrial cancer is ∼60%, exceeding that of colorectal cancer,23 24 25 26 while the risk of developing other extracolonic cancers ranges from 4–13%.24
MSI testing of tumours is a valuable tool in selecting cases with an underlying mismatch repair defect.27 One confounding factor in the identification of genuine HNPCC cases is their distinction from cases with sporadic colorectal cancers that also demonstrate MSI and loss of MLH1 expression. The latter occur predominantly in females over 65 years of age and are usually associated with biallelic methylation of the CpG island promoter of MLH1,28 29 and the presence of the BRAF V600E mutation.30 31 32 In contrast, HNPCC related cancers are invariably BRAF wild-type.30 31 32 On the basis of these findings, the molecular determination of BRAF mutation status is often used in clinical practice to select cases for germline testing.30 31 32 33 Immunohistochemistry (IHC) of the tumour plays a significant role in determining which gene within the mismatch repair system should be tested for a germline defect.34 35 36 Somatic loss of the wild-type allele in HNPCC associated tumours has been attributed to acquired deletions or chromosomal loss, point mutation, or promoter methylation in the case of MLH1.37 38 39 Genetic screening for germline mutations of the mismatch repair genes is usually conducted in peripheral blood DNA by a combination of direct sequence analysis of the exons to identify point mutations, and multiplex ligation dependent probe amplification (MLPA) in the case of MLH1 and MSH2, to detect large deletions or duplications common to these genes. Of all the mismatch repair mutations identified in HNPCC, mutations of MLH1 account for approximately 50% of cases, MSH2 for ∼40%, MSH6 for ∼10%, while PMS2 mutations are rare (2–5%).40 Nevertheless, mutations have collectively been identified in just 45–64% of families meeting the Amsterdam I criteria.7 18 41 Constitutional epimutations have been identified in a proportion of mutation-negative cases.8 9 10 11 12 13 14 15
Constitutional epimutation as an aetiological mechanism for HNPCC
An epimutation may be defined as an epigenetic aberration present in normal cells that causes transcriptional silencing of a gene that is normally active, or conversely, reactivation of a gene that is normally silent.42 43 Constitutional epimutations of MLH1 and MSH2 have been identified in a small number of individuals meeting the clinical criteria for HNPCC in whom no pathogenic sequence mutation of the mismatch repair genes was identified. Constitutional epimutations manifest as methylation of the cytosine bases within CpG dinucleotides across a single allele of the CpG island promoter and transcriptional silencing of the affected allele in normal tissues.
Constitutional epimutation of MSH2 in familial cases of HNPCC
Constitutional epimutations of MSH2 have been found in a handful of familial HNPCC cases in which multiple affected members had early-onset colorectal and endometrial tumours demonstrating MSI and dual loss of the MSH2 and MSH6 proteins, but no evidence of a germline mutation within the mismatch repair genes.16 17 In one family, in which the two alleles of the MSH2 promoter were distinguishable through the presence of heterozygous single nucleotide polymorphisms (SNPs), methylation was found to be confined to a single allele.16 However, the degree of allelic methylation was highly mosaic between tissues, varying from 0–3% in peripheral blood, to nearly 50% in epithelial tissues including colonic and buccal mucosa and endometrium.16 17 In each family, the MSH2 epimutation showed autosomal dominant transmission and segregated faithfully with a single allele of chromosome 2p21, implicating an underlying cis genetic defect.16 17 Deletions of the final exons of the EPCAM (formerly TACSTD1) gene, which is located immediately upstream of MSH2 and expressed in the same direction, were subsequently identified in the epimutation carriers. The deletions encompassed the transcription termination signal, abolishing transcriptional termination from EPCAM, and resulting in transcriptional read-through into MSH2. Notably, the fusion transcripts produced were elevated in epithelial tissues, where EPCAM expression is upregulated, correlating with the extensive MSH2 methylation observed therein.17 Thus, the HNPCC phenotype in these families was conferred by epimutations of an intact MSH2 gene, mediated by a failure in transcriptional termination from the neighbouring gene, which particularly affected epithelial tissues in which HNPCC associated tumours are most prevalent.
Constitutional epimutations of MLH1 and their molecular basis
The first case of a constitutional epimutation of MLH1 was identified in 2002 with the finding of extensive methylation affecting a single allele of the MLH1 promoter in the peripheral blood DNA of a sporadic proband with early onset colorectal cancer exhibiting MSI and loss of MLH1 expression.8 Deletion of the unmethylated allele in the tumour was detected by loss of heterozygosity (LOH).8 Constitutional MLH1 epimutations were further characterised with the identification of additional cases thereafter. In patients heterozygous for a common G>A SNP within the MLH1 promoter, methylation of CpG sites flanking the SNP was shown to be restricted to a single allele in each case (fig 1).9 10 11 12 Expressible SNPs located within an exon of the MLH1 gene, for which the epimutation carriers were heterozygous, were similarly exploited to trace the genetic allele from which the MLH1 mRNAs were transcribed. The observation of a single allele at the corresponding SNP site within the mRNA of normal cells confirmed transcriptional inactivation of the affected allele (fig 1).11 12

The molecular characteristics of a constitutional MLH1 epimutation. The molecular features of a constitutional MLH1 epimutation, as exemplified by “patient A”,12 are shown. Panel A: Schematic of the MLH1 gene showing the effect of a constitutional epimutation on it. The CpG island promoter is denoted by lollipops with methylated CpG sites in black, unmethylated CpG sites in white, and the informative single nucleotide polymorphism (SNP) within the promoter indicated. Exons are depicted as boxes and the expressible SNP site within exon 8 is shown. Transcriptional activity is indicated by a waved arrow and mRNA transcripts as waved rectangles. In “patient A”, methylation was specific to the A allele of the promoter SNP, and only the G allele at the exon 8 SNP was detected in the mRNA. Panel B: Clonal sodium bisulphite sequencing demonstrated that methylation was confined to the “A” allele at the promoter SNP. Following the treatment of genomic DNA with sodium bisulphite and subsequent polymerase chain reaction (PCR) amplification, unmethylated cytosines are converted to thymines via uracil, while any methylated cytosines within CpG dinucleotides are retained as cytosines. PCR products are cloned into a plasmid vector allowing individual alleles to be sequenced, as shown in each electropherogram. Methylation of the “A” allele (asterix) was demonstrated by the finding of cytosines at CpG sites flanking the SNP site. The “G” allele (asterix) was shown to be unmethylated by the finding of thymines at the corresponding CpG sites. Panel C: Monoallelic methylation of the “A” allele was found in all normal somatic tissues tested in “patient A”. CpG dinucleotides are depicted by circles, with black as methylated and white as unmethylated, and each horizontal line represents a single allele following clonal bisulphite sequencing. The promoter SNP is depicted by a box, with green as “A” and orange as “G”. Panel D: Sequence analysis across the expressible G/A SNP within MLH1 exon 8 showed that “patient A” was heterozygous (asterix), but only the “G” allele (asterix) was detected in her mRNA, indicating allelic transcriptional silencing of the “A” allele.
Constitutional MLH1 epimutations are likely to be associated with additional epigenetic changes that accompany cytosine methylation, including repressive histone modifications. Mono- or hemiallelic MLH1 methylation was found to be widespread throughout the normal somatic tissues of epimutation carriers, which included tissues derived from all three embryonic germ cell lineages; buccal and colonic epithelium (endodermal), peripheral blood lymphocytes (mesodermal), and hair follicles (ectodermal).9 10 11 12 This provided strong evidence that the epimutations, or the mechanism giving rise to them, are derived from the germline.10 Yet in the majority of cases there has been some degree of mosaicism in the level of allelic methylation present in the normal cells, with a proportion of epimutant alleles devoid of methylation, suggesting either incomplete penetrance or partial erasure of methylation in some cells. Pronounced mosaicism has been reported in a small number of cases, with methylation detected in as few as 10% of alleles, in concert with partial transcriptional repression of the affected allele.14 This novel epigenetic defect is quite distinct from the somatic epigenetic silencing of MLH1 in sporadic MSI colorectal cancers of the elderly, in which dense biallelic methylation of MLH1 and flanking genes is essentially confined to the cancer.44
Clinical presentation and management of cases with a constitutional MLH1 epimutation
Twenty-four confirmed index cases with a constitutional epimutation of MLH1 but without any sequence alteration of the gene have now been reported in the literature and in each proband the phenotype was consistent with HNPCC.8 9 10 11 12 13 14 15 Clinical data, including cancer type, age at diagnosis and family history of the probands, are summarised in table 1. The majority met at least one of the revised Bethesda guidelines for HNPCC, while a small number met stricter criteria due to the presence of HNPCC related cancer in at least one first degree relative. Approximately half of the cases presented with multiple primary tumours within the HNPCC spectrum. Clearly case reports are likely to be the subject of significant ascertainment bias and therefore do not allow conclusions regarding average age of onset of cancer in epimutation carriers. Having said this, it appears that cancer occurs at a young age in this group of patients, with a mean (SD) age of 37.3 (9.5) years for the first diagnosis of colorectal cancer in confirmed cases, comparable to individuals with sequence mutations of MLH1 (mean (SD) age 43.6 (9.9) years.36 45). Two additional carriers of an MLH1 epimutation have also been reported, who were a first degree relative of two of the probands, but were asymptomatic at the ages of 48 and 64 years.12 14 The molecular profile of tumours from individuals with constitutional MLH1 epimutations are similar to those found in individuals with conventional sequence mutations of MLH1. In epimutation carriers the tumours have demonstrated MSI and loss of the MLH1 protein (table 1). The “second hit” affecting the unmethylated allele was also identified in a proportion of these tumours (table 1), further indicating that the epimutation imparts the “first hit”, predisposing to the development of HNPCC-type cancers. In the small proportion of tumours screened for the BRAF V600E mutation, this was not found.14
Clinical features of reported cases with a constitutional MLH1 epimutation
A further five mutation negative probands have been reported who may also be carriers of a constitutional MLH1 epimutation (table 1).46 However, these cases require confirmation since the upstream region of the MLH1 promoter in which methylation was detected in these patients is also susceptible to methylation in normal somatic tissues in the normal population, without any detrimental effect on transcriptional activity.47 Furthermore, the screen was performed using a single methylation test, methylation specific PCR (MSP), which can be prone to false positive results.46 In two of these probands (H32, H46) the tumours demonstrated MSI, but had retained MLH1 expression,46 although it is possible that presence of an epimutation may not have been contributory to tumour development in these particular cases.
A single case with multiple tumours has recently been described with extensive allelic MLH1 promoter methylation in normal tissues in concurrence with a linked 6.4 kb deletion encompassing the transcription and translation start sites, as well as the first two exons of the gene (table 1).15 While this case may not strictly be classified as having a constitutional MLH1 epimutation, since the epimutation is accompanied by a sequence alteration within the affected gene, it may nevertheless provide crucial insight into the mechanism underlying the onset of classic MLH1 epimutations. In this case it is likely that methylation accrued on the affected allele as a secondary consequence of the loss of transcriptional activity incurred by the deletion.15
The currently available clinical data indicates that constitutional MLH1 epimutations confer a phenotype consistent with HNPCC, with a high risk of early development of HNPCC related cancers. On this basis, we suggest that individuals who carry a constitutional MLH1 epimutation should be offered the same risk management strategies as individuals with sequence mutations in the mismatch repair genes. In practical terms this means 1–2 yearly colonoscopies from age 25 years and the consideration of subtotal colectomy in selected patients.22 The salient difference in carriers of epimutations and their counterparts with sequence mutations relates to the risk of cancer in their family members, since epimutations do not conform to the classic autosomal dominant inheritance pattern seen in mutation carriers. There is a tangible risk that first degree relatives will have the condition, though at this stage, this appears to be <50%. However, given the recent case reports of epimutations in two generations, it is strongly advised that the first degree relatives of probands are counselled and offered molecular testing to determine their carrier status. One of the precautions with respect to screening for epimutations in a clinical setting relates to the possibility of false negative test results. This may occur because epimutations often show mosaicism between individuals and tissues. This possibility highlights a further distinction between the clinical interpretation of a predictive test for an epimutation compared with a sequence mutation. Predictive tests for sequence mutations can be used to confidently reassure unaffected clients and discharge them from high risk surveillance programmes. In our view it is inappropriate to adopt this strategy for family members who test negative for an epimutation. As discussed there is as yet an unquantified risk of false negative results and the implications of an inherited disease haplotype without detectable allelic methylation are unknown. Our practice is to recommend family members of epimutation carriers remain under surveillance until a clearer picture of the clinical profile, tumour characteristics and inheritance pattern of constitutional MLH1 epimutations emerges.
Non-Mendelian inheritance of constitutional MLH1 epimutations
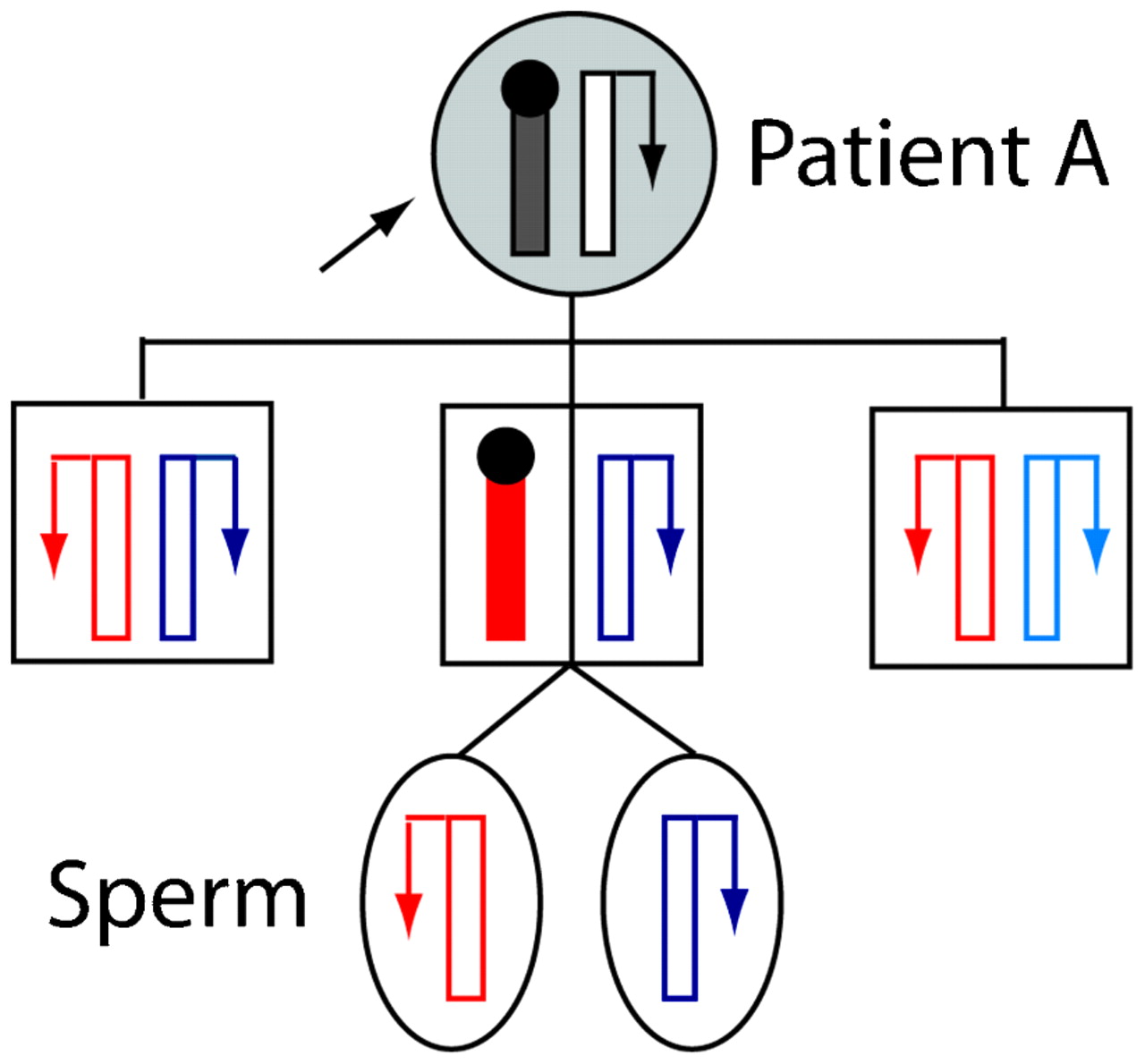
Most cases with a constitutional MLH1 epimutation have had no strong family history of HNPCC-type cancers. Coupled with the knowledge that epigenetic modifications are labile, this suggested that epimutations were unlikely to be transmissible from one generation to the next.48 The demonstration that the epimutation in an early sporadic case, “patient ST”, occurred de novo on the maternal allele supported this initial view.11 Nevertheless, the heritability of constitutional MLH1 epimutations has now been definitively demonstrated with the finding of intergenerational transmission of this defect in two familial cases.12 14 In the first reported case of “family A”, the constitutional MLH1 epimutation was transmitted in non-Mendelian fashion from an HNPCC affected female (patient A) to one of her sons, but was reversed in two other sons who had also inherited the affected maternal allele (fig 2).12 This intriguing finding was evidenced by the presence of soma-wide promoter methylation and transcriptional inactivation of the same MLH1 allele in the mother and one son, while in the other two sons the very same MLH1 allele was unmethylated and the gene biallelically expressed, indicating reversion of the affected allele to the normal functional state in them. Interestingly, the spermatozoa cells of the son carrying the epimutation were also devoid of methylation and the affected allele had resumed transcriptional activity. This indicated complete erasure of the epimutation in his germline, showing that while this defect is widespread in the soma, it is meiotically reversible (fig 2).12 This was consistent with the absence of MLH1 methylation detected in the spermatozoa of another male proband harbouring a constitutional MLH1 epimutation, “patient TT” (table 1).49 (Although ⩽1% methylation was detected in the first sample this proband provided,10 this was subsequently shown to be an artefact).49 In the second familial case of a constitutional MLH1 epimutation, a male HNPCC proband was found to have inherited the epimutation from his unaffected mother. Intriguingly, the MLH1 methylation was mosaic, affecting ∼10% of alleles in both the proband and his mother,14 suggesting the epimutation in this case was subject to somatic variation, possibly reflecting an underlying allelic susceptibility to methylation that might nevertheless be stably transmitted through the germline.

Non-Mendelian inheritance of a “reversible” constitutional MLH1 epimutation. Pedigree of “family A” showing stochastic maternal transmission of the meiotically reversible constitutional MLH1 epimutation in the different members.12 Alleles are depicted as vertical bars, with transcriptional activity indicated by an arrow, with methylation and transcriptional silencing by a black circle. Paternally inherited alleles are shown in different shades of blue, and alleles of unknown parental origin in black. The maternally inherited allele, which was genetically identical in each of the sons, is shown in red. Normally functioning alleles are in white and shaded alleles show presence of the epimutation. The proband “patient A”, who had presented with multiple hereditary non-polyposis colorectal cancer (HNPCC) related cancers, transmitted the epimutation to just one of her three sons, placing him at risk of developing HNPCC related cancers. However, the same allele reverted back to the unmethylated and fully functional state in the other two sons. The epimutation was also erased in the spermatozoa of the son, who bore the epimutation throughout his somatic tissues, indicating its epigenetic reversal in the male germline.
It is possible that the epimutations in different cases are caused by distinct mechanisms associated with different inheritance patterns and transmission risks, even though they each manifest methylation of the MLH1 promoter. It is notable that of the six index cases for whom familial DNA samples were available for analysis and genetically informative, the MLH1 epimutations either arose on the maternally derived allele, or were transmitted through the maternal germline. While it is tempting to speculate that this defect is transmissible solely via the maternal germline, it must be noted that no offspring of male probands have been tested for carrier status. While the epimutations are reversed in spermatozoa, this does not rule out the potential for transmission via the male germline if this defect is under the influence of a genetic factor. It remains possible that an epimutation could be reinstated in the somatic cells of the embryo post-fertilisation. The lack of knowledge regarding the mechanism underlying constitutional MLH1 epimutations, together with the non-Mendelian inheritance observed to date, present a genetic counselling conundrum. Until such a time as the true nature of this defect and its associated risk and pattern of inheritance are defined, molecular testing should be offered to all first degree relatives of probands with a confirmed MLH1 epimutation, with the caveat that a negative test result may not necessarily reflect their true risk of cancer development.
Triaged selection of patients for the molecular diagnosis of MLH1 epimutations
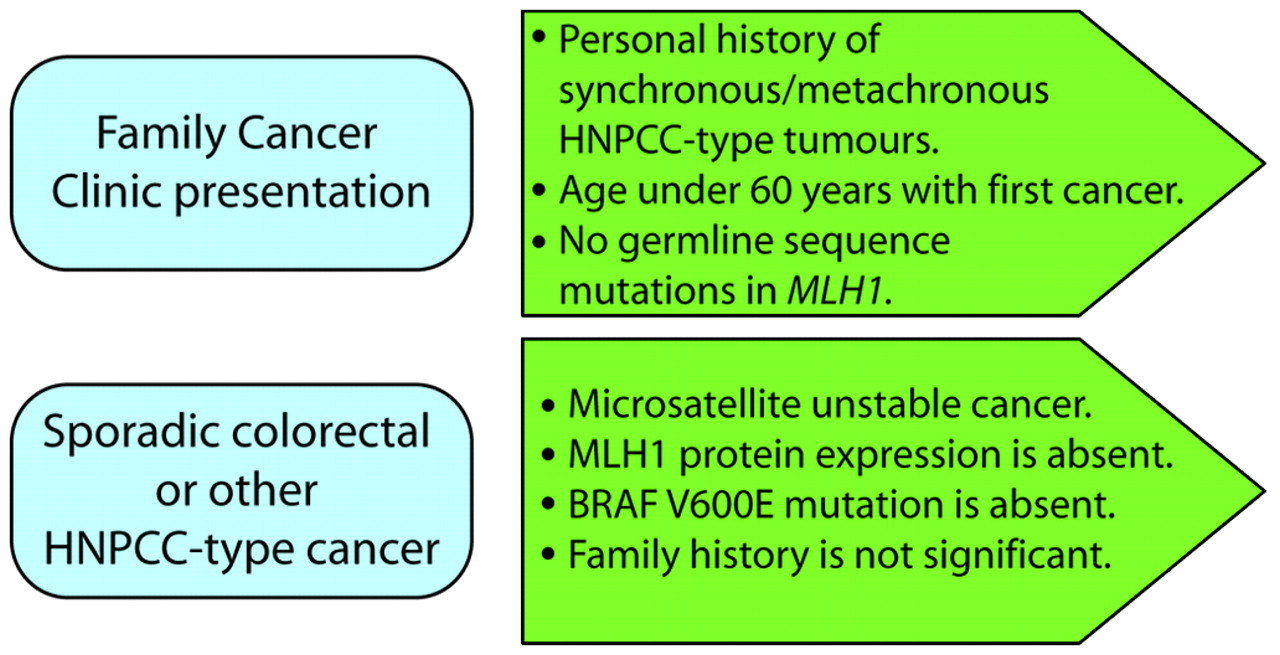
Constitutional MLH1 epimutations are rare among the HNPCC population, but their precise frequency has not been established as the various screens conducted to date have used different selection criteria. In one survey of 160 HNPCC probands selected on the basis that no pathogenic mutation of the MLH1 or MSH2 genes was identified, just one case was positive for a constitutional MLH1 epimutation.11 Constitutional epimutations were identified in about 10% of cases where the inclusion criteria employed were: (1) at least one of the revised Bethesda guidelines was met; (2) the tumour exhibited MSI and IHC loss of MLH1; and (3) no pathogenic mutation of MLH1 was found by standard genetic screening.9 12 13 Since constitutional MLH1 epimutations may confer a similar predisposition to the development of HNPCC related cancers as MLH1 sequence mutations, a molecular diagnosis is warranted so that carriers receive appropriate clinical surveillance and their relatives may be offered genetic counselling and molecular testing for carrier status. Given the increasing availability of molecular tumour testing, there is an opportunity to use information from the cancer to identify individuals who carry constitutional epimutations. Such individuals may present with sporadic cancer or as clients of family cancer clinics. Certainly, the young age of onset of cancer may have prompted referral to a family cancer clinic without a significant family history of colorectal or other cancers. We propose that individuals who have undergone germline sequencing of MLH1 and in whom no sequence mutations have been identified should be screened for constitutional epimutations in this gene (fig 3). Furthermore we suggest that individuals who present with a sporadic colorectal cancer which is MSI, fails to express MLH1 and does not display a BRAF V600E mutation should also be considered for an epimutation screen (fig 3). The impetus to undertake such screening would be particularly strong if the individual had a personal history of synchronous or metachronous cancers and was under the age of 60 years at first diagnosis. If the individual reported a strong family history of cancer, then germline sequence mutation testing should be prioritised before epimutation screening.

{kind=link}
{kind=link}
{kind=link}
Presentations prompting consideration of screening for a constitutional MLH1 epimutation. HNPCC, hereditary non-polyposis colorectal cancer.
Molecular diagnosis of constitutional MLH1 epimutations
Currently the most definitive test for the molecular diagnosis of constitutional MLH1 epimutations is the detection of allelic methylation at the MLH1 promoter in the constitutional DNA. There is presently no “gold standard” test for this, however, there are two key considerations to take into account. Firstly, the assay employed for the initial detection of methylation should target the region of the MLH1 promoter between the transcriptional start site and 250 bp upstream, encompassing the “Deng C” and “Deng D” regions, since this segment best predicts loss of transcription and is less susceptible to non-specific methylation.47 Secondly, each technique has its limitations and so the application of more than one screening method is recommended. Certainly the finding of a positive result in constitutional DNA with an initial assay would require confirmation of the presence of allelic methylation using a secondary method.
Most techniques currently employed to detect the presence of methylation are based on the treatment of genomic DNA with sodium bisulphite, which then serves as the template for a subsequent PCR based assay. Sodium bisulphite converts unmethylated cytosines to uracils, and thence to thymine during PCR amplification, whereas methylated cytosines are unreactive.50 Thus, methylation is detected by the retention of cytosines at CpG dinucleotides within the CpG-rich promoter region, as opposed to thymines at the corresponding sites when the promoter is unmethylated (fig 1B). PCR based assays for the detection of methylation are designed to exploit this sequence difference. These include traditional combined bisulphite and restriction analysis (COBRA), which employs restriction endonucleases that contain a differential CpG within their cleavage recognition site.51 MSP and MethyLight make use of allele specific oligonucleotides designed to specifically amplify templates that have retained the cytosines at CpG sites.52 The latter methods are highly sensitive, as well as quantitative when performed with fluorescent labels against a standard curve.12 49 52 However, MSP is prone to bias and false positive results unless carefully optimised. Pyrosequencing measures the relative levels of cytosine and thymine bases at designated CpG sites within a short stretch of sequence.53 Methylation specific MLPA (MS-MLPA) is an alternative technique that has been successfully implemented for the identification of MLH1 epimutations.14 15 MS-MLPA does not require prior sodium bisulphite conversion of DNA, but instead makes use of methylation sensitive restriction endonucleases that fail to cleave the (CpG-containing) recognition site if it is (hemi-)methylated.54 In MS-MLPA, probes are designed to encompass such a restriction site and anneal to the denatured genomic DNA. Unmethylated DNA is digested and thus fails to amplify, whereas hemimethylation DNA probe hybrids result in a product. The relative level of methylation is detected in the digested DNA versus an undigested sample.54 55 56 MS-MLPA presents a reliable semi-quantitative technique suitable for a first pass screen for MLH1 epimutations, especially for laboratories practised in traditional MLPA but unfamiliar with sodium bisulphite based methods. Each of the above techniques, as applied to the MLH1 gene, can readily detect the presence of 10–50% allelic methylation in constitutional DNA. Allelic bisulphite sequencing or the demonstration of allelic transcriptional downregulation at an informative SNP site provides clear confirmation of the presence of a constitutional MLH1 epimutation (fig 1).
Potential mechanisms for the onset and inheritance of constitutional MLH1 epimutations
The mechanism by which constitutional MLH1 epimutations arise and are transmitted to a proportion of offspring, but reversed in others, remains to be elucidated. MLH1 epimutations may have a fundamental epigenetic basis or may result from predisposing genetic factors. It appears that whatever the underlying error is, it is likely to occur more frequently in the female germline. If constitutional MLH1 epimutations originate from a spontaneous epigenetic aberration, transmission from the carrier to future generations would seem unlikely as this would require that the epimutation be transmitted intact (with epigenetic modifications still attached to the affected allele) through the germline to the offspring in a process referred to as “gametic epigenetic inheritance”.57 The major epigenetic reprogramming events that occur at various stages of the mammalian reproductive lifecycle would present a significant hurdle to this, as follows. During gametogenesis, parental origin epigenetic tags are established at imprinted genes to reflect the sex of the parent.58 It is plausible that an epimutation might arise at this point such that the epigenetic modifications are in place within the gamete. Following fertilisation, the paternal genome encased within the male pronucleus of the zygote is subject to rapid demethylation.59 Further global demethylation of the genome accompanies cell division in the preimplantation embryo, until somatic methylation patterns are established de novo in the inner cell mass of the blastocyst.58 Imprinted genes withstand these post-fertilisation epigenetic fluctuations,58 and it is possible that a spontaneous epigenetic aberration that originated in the gamete might similarly do so, ultimately becoming disseminated throughout the somatic tissues of the offspring. However, the epigenetic marks at imprinted genes are completely erased in the primordial germ cells (the precursors of the developing fetus’ future gametes) during their migration down the genital ridge.58 One would anticipate that a spontaneous epigenetic error would be eradicated contemporaneously at this stage.60
To date, gametic epigenetic inheritance in mammals has only been demonstrated definitively in inbred mouse strains (Avy and AxinFu) in which the phenotype is driven by ectopic expression derived from the promoters of nearby intracisternal A particle (IAP) retro-elements.57 Gametic epigenetic inheritance in these mice is likely attributable to the resistance of this type of sequence element to epigenetic reprogramming in primordial germ cells.61 Following an initial claim that MLH1 methylation was retained in a small proportion (<1%) of the spermatozoa of a male proband (patient TT, table 1), it was suggested that gametic inheritance of the epimutation could occur, albeit at low risk.10 However, subsequent re-evaluation of the original spermatozoa sample, in which a control for somatic contamination was incorporated, indicated that the small trace of MLH1 methylation initially reported was an artefact equivalent to the amount of contaminating somatic DNA. No methylation was detected in a second sample of spermatozoa donated by proband TT.49 Thus the efficient eradication of the constitutional MLH1 epimutations in the spermatozoa of two male carriers now argues against a role for gametic epigenetic inheritance via the male germline.49 Certainly the chromatin remodelling that occurs during spermiogenesis, which includes the replacement of the majority of histones with protamines, renders this mode of transmission highly unlikely.62 However, the propensity for a failure in epigenetic resetting in the female germline remains obscure and the potential for maternal gametic epigenetic inheritance is unprecedented in humans. Most studies of mammalian epigenetic reprogramming have been conducted in mice, which may not be entirely representative of the events that occur in humans.
Constitutional epimutations may arise following a genetic cue. The majority of epimutations reported for other genes have been caused by genetic alterations acting in cis. For example, in a case with α-thalassaemia, promoter methylation and transcriptional silencing of the HBA2 gene was mediated by antisense transcription upstream through the gene. This was initiated from the promoter of the LUC7L gene downstream, which had been truncated by an interstitial deletion.63 In families with a predisposition to B cell chronic lymphocytic leukaemia, promoter methylation and transcriptional suppression of the affected allele of the DAPK1 gene was attributable to a point mutation upstream of the DAPK1 promoter that resulted in a higher binding affinity for the HOXB7 transcriptional repressor.64 Autosomal constitutional epimutations caused by a linked, fully penetrant genetic defect would be expected to conform to a predictable Mendelian pattern of inheritance, as exemplified by MSH2 epimutations.17 With the notable exception of one case,15 no genetic alterations within the MLH1 gene have been identified in individuals harbouring a constitutional MLH1 epimutation. Furthermore, there is no evidence to support a cis-acting genetic defect underlying reversible MLH1 epimutations. In family A, in which the epimutation was present in the mother and one son, but had been reversed in the other two sons (fig 2), there was no discernible genetic difference in the affected allele in these four family members, irrespective of whether the epimutation was present or reverted. Furthermore, haplotyping across megabases flanking MLH1 found no evidence for any recombinations in the vicinity of MLH1 in any of the children. Therefore it is unlikely that the epimutation in this particular family was caused by a linked genetic defect, unless any such defect is associated with incomplete penetrance.12
An unlinked genetic defect that acts on MLH1 in trans could allow for the intergenerational transmission of an epimutation to a proportion of offspring in which the putative genetic defect was co-inherited. For example, co-segregation of a mutant epigenetic modifier during meiosis could impart a proportion of gametes with an epimutation (or subsequent propensity to it). In this scenario, the epimutation need not be maintained with its epigenetic modifications intact within the germline itself, but could be re-established post-fertilisation in the subsequent generation. This has been shown to occur in Drosophila.65 If this proves to be the case, constitutional MLH1 epimutations may be transmissible by either parent and the respective epigenetic states in gametes would provide no indication of inheritance risk. Alternatively, the trans-acting factor might be specific to the maternal germline, giving rise to an epimutation in a proportion of oocytes, consistent with the maternal bias in origin of constitutional MLH1 epimutations. Irrespective of this, the effects exerted by a mutant trans-acting epigenetic modifier are unlikely to be restricted to MLH1. While no altered epigenetic states were observed at a small number of selected genes in cases harbouring a constitutional MLH1 epimutation,10 11 the possibility of more disseminated epigenetic disruption has not been comprehensively investigated. It is plausible that other genes are targeted, with methylation similarly confined to a single allele. Concordant epimutations of additional loci may not be associated with any apparent phenotype provided the normally functioning allele was retained (unless any such phenotype is associated with haploinsufficiency). Nevertheless, the possibility of aberrant methylation of additional genes in cases harbouring an MLH1 epimutation warrants genome-wide investigation, not only to determine the potential role for a trans-acting epigenetic regulator in its establishment and inheritance, but also because the potential unmasking of epimutations at other loci could modify the phenotype.
The major determinant of the risk and pattern of inheritance conferred by a constitutional MLH1 epimutation is the nature of the mechanism underlying it. Elucidation of this mechanism has significant clinical implications, both in terms of diagnostic screening protocols and ascertaining an accurate risk of inheritance for genetic counselling purposes. However, these issues could be further complicated if MLH1 epimutations differ in their underlying cause between particular individuals and families. Further clarification of this defect is clearly required before the full ramifications of its aetiological role and in disease causation and risk of transmission to offspring are realised.
Conclusion
There has been some debate over the appropriate naming of this defect. The term “germline epimutation” was conceived on the basis that the aberrant promoter methylation affected a single parental allele, was present in tissues derived from all three embryonic germ cell lineages, and initially thought to be present in spermatozoa.10 While we know that this defect can be passed from one generation to the next, and therefore the mechanism that underlies it is transmissible through the germline, there is currently no definitive evidence to demonstrate that the epimutation is borne autonomously within the gamete. It may be the case that epimutations are reversed in the gametes and re-established in the somatic cells in successive generations. The general consensus is that the term “constitutional epimutation” is more appropriate, as it indicates the presence of an epigenetic aberration in normal somatic cells to underlie a disease condition, but neither precludes nor dictates that its origin is in the germline, or that it is distributed evenly throughout the soma. We have adopted the broader term “constitutional epimutation” as it embodies any epimutation that affects a single allele of a gene (of normal sequence) irrespective of its origin or mechanism, and recommend its use henceforth.
REFERENCES
Footnotes
Funding MPH is supported by a Career Development and Support Fellowship from the Cancer Institute NSW and research funds from the Australian NH&MRC.
Competing interests None.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.