Article Text

Abstract
The relationship between paroxysmal movement disorders (PD: paroxysmal dyskinesia) and epilepsy continues to present a challenging problem. Attacks of PD and epileptic seizures have several characteristics in common: both are paroxysmal in nature with a tendency to spontaneous remission, and a subset of PD responds well to anticonvulsants. In 1997, description of the ICCA (infantile convulsions and choreoathetosis) syndrome and linkage to chromosome 16p12-q12 provided the first genetic evidence for common mechanisms shared by benign infantile seizures and PD. The chromosome 16 ICCA locus is by far the most frequently involved in such associations as well as in pure forms of benign infantile seizures. The ICCA region at the pericentromeric area of chromosome 16 shows complicated genomic architecture and the ICCA gene still remains unknown. Genetic studies focusing on PD with or without epilepsy have led to the identification of other genes linked to chromosomes 2q35 and 10q22. Alterations of ion channel and ion pump subunits could provide a simple, albeit probably non-unique, explanation for the pathophysiology of the link between epilepsy and PD. The aim of this review is to update genetic aspects of infantile epileptic seizures and PD and their association in the context of ICCA and ICCA related syndromes.
Statistics from Altmetric.com
The human epilepsies can be broadly divided into two main categories: the symptomatic epilepsies that arise from separate brain lesions or disorders; and the idiopathic epilepsies, which have a strong genetic component. A large number of distinct epileptic syndromes can be defined according to the international classification of epilepsy syndromes,1 and as much as 30% of all the epilepsies may be of genetic origin.2 Most forms of human idiopathic epilepsy show complex mode of inheritance: several genes interacting with each other and/or with environmental factors contribute to the pathophysiology of the disease.3 4 In contrast, rare epileptic syndromes may be inherited as monogenic traits in some families. So far, most, if not all, known idiopathic epilepsy genes have been identified in those epileptic disorders that display Mendelian inheritance.5 The majority of those genes encode ligand or voltage gated ion channel subunits,6 7 which is not surprising given the crucial role of such ion channels in neuronal functioning. Ion channels may well represent common targets of molecular alterations shared by inherited and acquired epilepsies.2
BENIGN INFANTILE SEIZURES
Benign familial infantile seizures (BFIS) represent one of such epileptic syndromes that can be inherited as Mendelian traits. BFIS (MIM 601764, 605751, 607745) correspond to non-febrile convulsions with the first seizure at age 3–12 months and a favourable outcome with a normal psychomotor development. Typical seizures are brief with motor arrest, deviation of the head and eyes to one side, generalised hypertonia, cyanosis, and limb jerks. Seizures are focal with or without secondary generalisation and usually occur in clusters. In most cases the interictal electroencephalogram (EEG) is normal while the ictal EEG shows abnormalities that may originate from various cerebral lobes. In addition to the familial forms, non-familial cases have been described.8 They both belong to the group of so-called benign infantile seizures (BIS) according to the international classification.1
PAROXYSMAL DYSKINESIA
Paroxysmal dyskinesia (PD) represents a rare and heterogeneous group of neurological movement disorders. They are characterised by episodes of involuntary abnormal movements (dyskinesia) that correspond to attacks of choreoathetosis or dystonia. Very early descriptions of movement triggered attacks of choreoathetosis or dystonia have been reported.9–12 In a few cases, PD may be secondary to various causes including multiple sclerosis, hypoxic encephalopathy, or hypoparathyroidism.13 However, most PD appear idiopathic and are inherited as monogenic traits (see below). It has long been usual to distinguish between paroxysmal kinesigenic dystonia/choreoathetosis and non-kinesigenic paroxysmal dystonic choreoathetosis. A more recent classification14 divides PD into three main categories: kinesigenic PD (PKD), exercise induced PD (PED), and non-kinesigenic PD (PNKD).
PKD (MIM 128200) is characterised by sudden episodes of involuntary movement attacks. Sporadic and familial cases have been described. Age at onset is usually between 1 and 20 years, although this may vary especially in familial cases. The attacks are short in duration (<5 min) and are usually precipitated by sudden voluntary movements, but they may also be triggered by startling occurrences, hyperventilation, and continuous exercise. There is no loss of consciousness and response to antiepileptic drugs is usually good. In PED, dystonic attacks mainly of the legs last usually >5 min and occur after prolonged exercise; they are not precipitated by the classical PKD and PNKD triggering factors. PNKD (MIM 118800)—also known as paroxysmal dystonic choreoathetosis or Mount–Reback syndrome—is an autosomal dominant disorder characterised by spontaneous hyperkinetic attacks that usually last several hours and can be triggered by a large variety of stimuli, including fatigue, stress, menstruation, coffee and alcohol.
PAROXYSMAL DYSKINESIA AND EPILEPSY
The epileptic origin of PD has long been a matter of debate15–18 and PDs have even been classified as reflex epilepsies.16 EEG abnormalities have also been detected in a few patients with PD.19 20 The association of BIS with PD is particularly striking and has frequently been described in the same patients21 and in the same families.15 22–28 Less frequently, non-infantile epileptic seizures may also associate with PD; indeed, idiopathic generalised epilepsy,29 and in particular absence epilepsy,30–32 or even more complex epileptic phenotypes33 can be co-inherited with PD.
The pathophysiology of PD remains largely unknown. Most but not all PDs (see below the case of the MR1 gene) may well belong to the group of the so-called “channelopathies” in keeping with various types of paroxysmal cerebral and non-cerebral neurological syndromes, including the epilepsies.33–35 PDs have even been considered as subcortical epilepsies and the involvement of the thalamus or the basal ganglia has been proposed.36 Abnormal cortical and spinal functioning have also been detected.37
EPILEPSY AT THE CROSSROADS
The epilepsies are situated at the crossroads between various cerebral disorders, whether paroxysmal or not. Common clinical features, epidemiological studies, shared therapeutic strategies, and familial co-inheritance all argue in favour of close relationships between various epileptic syndromes and other paroxysmal disorders of the brain,35 such as migraine, episodic ataxia and, as mentioned above, PD.38 Similarly, the existence of links between the epilepsies and non-paroxysmal brain disorders such as autism, mental retardation and language impairment are well established. Genetic studies of the rare families where such epileptic and non-epileptic disorders are co-inherited as single Mendelian traits, present crossroads for the understanding of these clinically distinct disorders and their close relationships. Examples include: mutations of the KCNA1 potassium channel subunit gene in epilepsy with episodic ataxia type 139; rare mutations within the ATP1A2 sodium/potassium pump subunit gene in familial hemiplegic migraine, associated or not with infantile convulsions40 41 (see also below); the murine gene encoding the Cacna1a calcium channel subunit which is mutated in models of absence epilepsy,42 while its ortholog CACNA1A may be associated with epilepsy,43 hemiplegic migraine44 or episodic ataxia45 in humans; the X-linked SRPX2 gene that codes for a sushi-repeat protein of unknown function which is responsible for rolandic epileptic seizures associated with oral and speech dyspraxia46; and, in 1997, description of the ICCA syndrome (infantile convulsions and choreoathetosis; MIM 602066) and linkage to chromosome 16 (see below) which provided the first genetic evidence for common mechanisms shared by BFIS and PD.22
GENETICS OF BENIGN INFANTILE SEIZURES
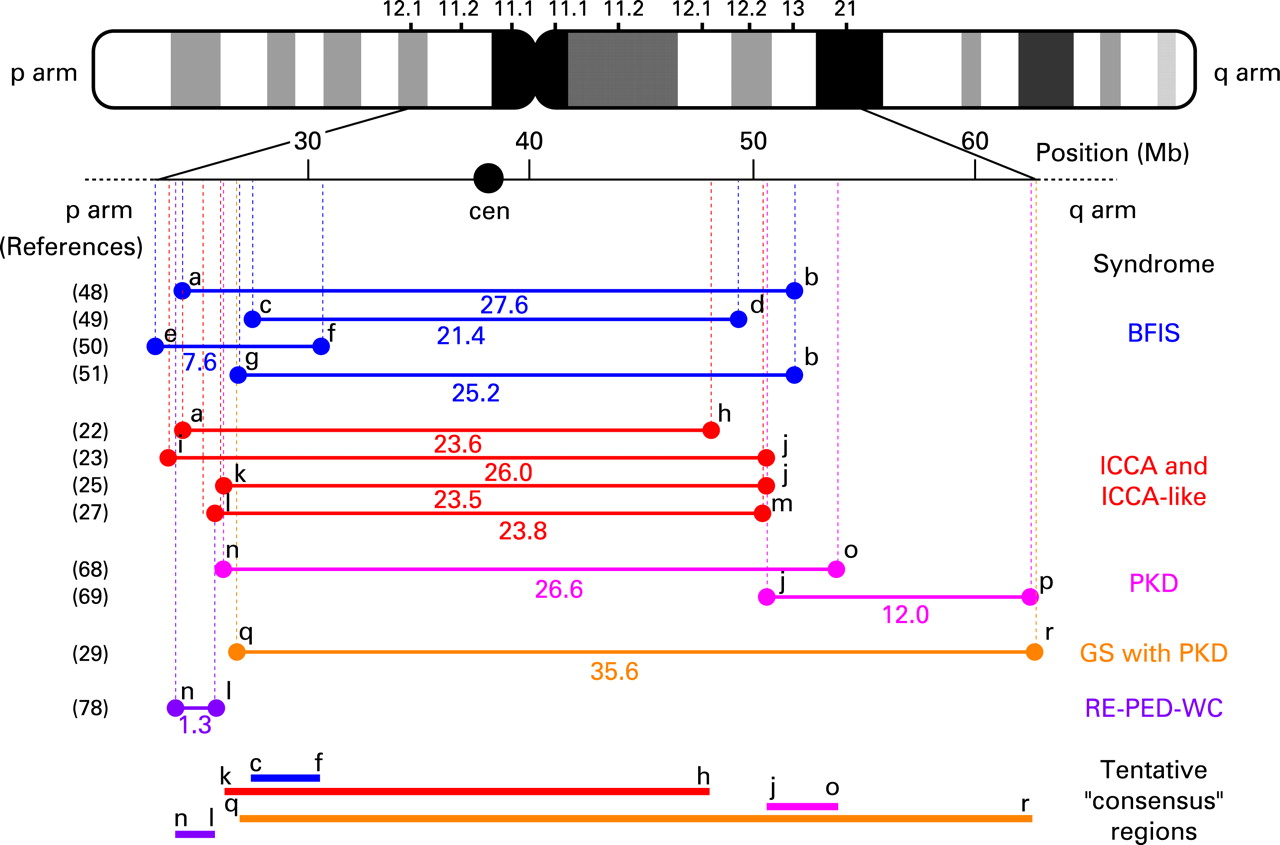
When familial, BIS is an autosomal dominant disorder with incomplete penetrance. Genetic heterogeneity has been demonstrated and several BFIS genes have been mapped (fig 1) at chromosome 19q in five Italian families,47 chromosome 16p12-q12 in more than 30 families worldwide,48–51 and chromosome 2q24 in four additional Italian families.52 The locus at chromosome 16 seems to be by far the most frequently linked. This latter point is also emphasised by the major involvement of the 16p12-q12 locus in those BFIS that are associated with PD (ICCA syndrome; see below) (fig 2). Exclusion of linkage to the three BFIS loci mentioned above suggested the existence of a fourth BFIS locus in an atypical form of BFIS with slight magnetic resonance imaging abnormalities of the brain.53


{kind=link}
{kind=link}
As already mentioned, the ATP1A2 gene situated at chromosome 1q23 is mutated in familial hemiplegic migraine (FHM).40 54 This gene codes for the α-2 subunit of a sodium/potassium ATPase. Co-segregation of BFIS with FHM in one Dutch-Canadian family led to the identification of a p.R689Q missense mutation in ATP1A2.41 p.R689Q occurs in the large intracellular catalytic loop between transmembrane domains M4 and M5. In vitro study of the mutation in HeLa cells revealed a decrease in the catalytic turnover and an increase in the apparent affinity for potassium ions with no change in ATP affinity, resulting in lower molecular activity of the α-2 pump isoform and a delay in extracellular K+ clearance.55 In contrast with these findings, no mutation in ATP1A2 has been found in families with pure BFIS to date,56 suggesting that BFIS with FHM, and BFIS alone, are genetically distinct. Additional genetic heterogeneity of BFIS is indicated by genetic exclusion of 1q23, 2q24, 16p12-q12 and 19q in two more BFIS families.50
Benign familial neonatal convulsions (BFNC; MIM121200, 121201) represent an autosomal dominant epileptic syndrome distinct from BFIS and can be caused by mutations in the genes encoding the KCNQ2 or the KCNQ3 potassium channel subunits.57–59 A few patients of families with typical BFNC may have epileptic seizures with semiology and age of onset consistent with the diagnosis of BIS.59–61 Those peculiar “BIS” patients inherited the same KCNQ2 or KCNQ3 mutations as their BFNC relatives, making it possible that the BIS phenotype in these BNFC families is actually due to defects in the KCNQ2/KCNQ3 tetrameric channel. Higher degree of complexity might exist. A clinical entity with the different, albeit related, phenotype (age at onset intermediate between BFNC and BFIS) of benign familial neonatal infantile seizures (BFNIS; MIM607745) has been defined.62 Mutations have been found within the SCN2A gene located at chromosome 2q24 and coding for the α-2 subunit of the voltage gated sodium channel subunit63 64 (fig 1). To date, nine different disease causing missense mutations have been identified in SCN2A. The mutant amino acids are located either in the transmembrane domains or in the intracellular loops of the protein. For this reason, a unified effect on protein function cannot be predicted; furthermore, no functional study on these mutants has been reported so far. A recent study showed that single BIS individuals within two novel BFNIS families did not inherit the SCN2A causing mutations65 and represented phenocopies, thus arguing against the existence of a molecular overlap between BFIS and BFNIS. The situation remains unclear; while a novel SCN2A mutation has indeed been found in a single and apparently typical BFIS family,66 no mutation within SCN2A has been detected in a series of 10 typical BFIS families.64 Moreover, whether SCN2A mutations are responsible for those BFIS that had been linked to 2q24 in four Italian families52 remains unknown to date. In this latter report, age at onset looked more consistent with the diagnosis of BFNIS than that of BFIS64; however, mutations in SCN2A are unlikely as this gene is situated just outside the 0.7 cM critical area—but phenotyping or genotyping errors cannot be firmly excluded.
GENETICS OF PAROXYSMAL DYSKINESIA
Genetics of PKD
Despite the existence of several PKD genetic loci (fig 1), no PKD gene has been identified so far. A peculiar form of autosomal dominant PKD with episodic ataxia and spasticity has been mapped at chromosome 1p.67 Another PKD locus (PKC1) has been mapped at the ICCA locus (see below) onto chromosome 16 in one large Afro-Caribean family68 (fig 2). Several so-called “PKD” families have been mapped to this PKC1 locus25 27; however, most PKD patients in these latter families also showed BIS so that the families should better be considered as ICCA or ICCA-like ones. A second locus (EKD2) has been defined at chromosome 16 in a PKD family of Indian origin69 (fig 2).
Genetics of PNKD
PNKD is autosomal dominant and genetic linkage has been obtained at chromosome 2q33-q3670 71 (fig 1). While this disorder was suspected to be a channelopathy, mutations in the non-ion channel gene MR1 have been found in several families.72–75 MR1 encodes a myofibrillogenesis regulator protein of unknown function and is expressed in structures of the brain critically involved in motor function such as the cerebellum, the spinal cord and the basal ganglia. Three MR1 isoforms (L, M and S) with various subcellular localisations have been described.72 Only two recurrent missense mutations (p.A7V and p.A9V) have been detected so far. Family history and haplotype analyses indicated that a founder effect is not responsible for the majority of the two recurrent mutations. Both mutations are situated within the same N-terminal α helix of the L and S isoforms of the MR1 protein—a part that is lacking in isoform M. Hence, it has been postulated that this helix probably plays an important role in the proper function of the L and S isoforms. The pathophysiological mechanisms associated with MR1 defects remain unknown. Coffee and alcohol are well known triggering factors in PNKD, both containing high levels of methylglyoxal. The MR1 protein is homologous to the HAGH (hydroxyacylglutathione hydrolase) protein that is part of the glyoxalase system, which detoxifies methylglyoxal. However, whether MR1 plays a similar detoxifying function remains undetermined to date. MR1 mutations may account for the majority of familial PNKD but there is genetic heterogeneity in PNKD.76 Interestingly, a recent study on phenotype–genotype correlation revealed clinical discrepancies between the PNKD families with MR1 mutations, and those without MR1 mutations.77
CLINICAL DESCRIPTION OF THE INFANTILE CONVULSIONS AND CHOREOATHETOSIS SYNDROME
The specific and familial association of BIS with PD in four families from northwestern France led us to define a novel clinical entity: the infantile convulsions and choreoathetosis syndrome (ICCA) (MIM 602066)22; earlier case reports15 21 were consistent with our definition.
In 1996, one of us (JR) re-examined and interviewed 83 adults and children from four different families living in the same part of France and possibly related, the families spanning more than four generations. All four families had previous histories of convulsion previously diagnosed in the first year of life as BFIS with apparent autosomal dominant inheritance and incomplete penetrance.
The first observation was made in a family spanning over three generations and comprising about 50 family members (family A).22 A 7-year-old girl who was initially diagnosed with partial seizure and secondary generalisation at 4 months of age was the first re-examined case. At 7 years she developed jerky movements of the legs after running >100 m when she had to stop fearing she would fall. She also presented with twisting movements of the hands when stressed or embarrassed—for example, when being asked a question in school or when asked to go to the blackboard in class. Several times we witnessed mild choreoathetotic movements of both upper extremities and slow athetoid posturing of both lower extremities. Initially, abnormal movements were intermediate in speed between quick and slow, typical of paroxysmal choreoathetosis. The arm would adduct and the elbow flex, followed by spastic posturing of the forearm and hand; the knee would flex, the ankle pantarflex and foot invert. Combinations of abnormal movements involving the arms, legs, trunk and occasionally the head were observed. The closest picture we could compare to in the literature was that of case 2 in Kinast et al.17 In our case, the attacks lasted only a few minutes, occurring with a frequency of 5–30 episodes per day, and were not accompanied by unconsciousness.
A younger brother (6 years of age) who was also affected with BFIS displayed exactly the same abnormal movements except that movements of the legs were more jerking. The mother who had had no seizure in her infancy displayed intermittent episodes of muscle spasms starting in one arm or one leg and spreading to the other when younger. These episodes were described soon after choreoatheoid posturing of both upper extremities and facial grimacing. At rest, she would first experience brief premonitory sensations of “movement” in both shoulders. Attacks were not severe, lasting for a maximum of 1 min. Attacks disappeared at 26 years of age without any treatment.
Typical choreoathetotic attacks are unprovoked by movements, whereas pure paroxysmal kinesigenic choreoathetosis is characterised by brief attacks of involuntary movement precipitated by the sudden onset of movement. In family A, attacks could happen in any situation: at rest, for example, when watching television, under emotional stress or after a brief exercise ranging from fast walking to standing up quickly from a sitting position. Between attacks neurological examination was entirely normal. Psychomotor development was not affected in all patients. Migraine was not present. We could observe the same type of abnormal movements in the three other families (families B–D)22 for individuals whether previously affected with BFIS or not. There was no specific abnormal movement in individuals affected with previous BFIS compared to those not previously affected. While some degree of intrafamilial heterogeneity was noticed, no movement abnormality was specific to any given family.
GENETIC EVIDENCE FOR SHARED MECHANISMS BETWEEN BENIGN INFANTILE SEIZURES AND PAROXYSMAL DYSKINESIA: THE ICCA AND ICCA RELATED SYNDROMES
It had long been postulated that mutations in ion channel genes could represent the pathophysiological link between epilepsy and PD. In support of this link, a missense mutation in the KCNMA1 gene that codes for the α pore-forming subunit of the large conductance (maxi K), calcium activated potassium channel and that is located onto human chromosome 10, has been found in a single family in which absence epilepsy and PD co-segregated32 (fig 1). This channel is activated by both membrane depolarisation and a rise in cytosolic Ca2+ concentration. The mutation (p.D434G) is located in the RCK (regulator of conductance for K+) domain, which may contain binding sites for a variety of regulatory ligands, including Ca2+ and Mg2+. Expression of the mutant protein in Xenopus laevis oocytes and in mammalian CHO cells showed an increase in open channel probability due to a three- to fivefold increase in Ca2+ sensitivity. This could lead to a gain of function of the α-subunit by inducing rapid repolarisation of action potentials, leading to increased excitability. It is noteworthy that a recent mutation screen of 18 families with various epileptic syndromes (16 with BIS, one with absence epilepsy, and one with generalised tonic–clonic seizures only) associated with PD did not detect any KCNMA1 mutation, at least within the coding sequence of the gene (unpublished data).
As mentioned above, the ICCA syndrome is characterised by the specific association of BIS with PD. It is inherited as an autosomal dominant trait with incomplete penetrance and phenotypic variability. Linkage of the ICCA syndrome to the pericentromeric area of human chromosome 1622 represented the first genetic evidence for shared mechanisms between BIS and PD. Since then, several studies have confirmed linkage at chromosome 1623–25 27 38 (fig 1 and 2). The majority of pure BFIS may well be allelic to ICCA, as linkage to the ICCA locus has been demonstrated in numerous families with BFIS only.48–50 Interestingly, a particular patient in one BFIS family had both BFIS and early onset PD.48 While all his affected relatives were heterozygous for the mutant allele and had typical BIFS only, this patient had inherited two copies of the disease haplotype. The related syndrome of autosomal recessive rolandic epilepsy with paroxysmal dystonia and writer’s cramp (RE-PED-WC; MIM 608105) has also been linked to the ICCA locus in one Sardinian family78 (fig 2).
While numerous studies have demonstrated significant linkage of various related syndromes including ICCA, ICCA-like and BFIS, to chromosome 16p12-q12 (fig 1), the disease gene(s) still remain(s) unidentified. A large number of genes lie within the critical area(s) that could be defined (see the human genome sequence website at http://genome.ucsc.edu) (fig 2). We (see Roll et al79 as an example) and others12 have screened numerous genes with no mutation found, but many genes remain to be analysed. Moreover, the ICCA region has a very complex genomic structure with highly duplicated large DNA sequences80 81 presenting an enormous challenge for mutation search. While the BIS, PKD and ICCA syndromes that are linked to 16p12-q12 (fig 1) may well be allelic, the existence of several homologous disease genes in this particular chromosomal region is not unlikely as nearly identical copies of genes might exist in the ICCA region. For example, four genes encoding members of the sulfotransferase family (SULT1A1-4) are located on and among duplicated low copy repeats at chromosome 16p11.2,82 and the SULT1A3 and SULT1A4 copies even seem to have undergone further intra-chromosomal duplication (UCSC genome browser on human March 2006 assembly: http://genome.ucsc.edu/cgi-bin/hgTracks; position: chr16:28,500,000–30,144,000). The determination of a single and consensus critical region of interest (fig 2) would be hampered and could be associated with dramatic errors if more than one disease gene in the ICCA region caused identical or closely related disorders. Moreover, artefacts of wrong genome assembly remain possible.
In the context of such a complicated genomic architecture, the occurrence of large scale genomic rearrangements such as deletions, duplications and inversions would be favoured and hence cannot be excluded. Indeed, deletions at 16p12-p11 have recently been detected in four patients with sporadic microdeletion syndrome consisting of developmental disabilities,83 but none of the four patients had ever shown epileptic seizures or abnormal involuntary movements. Very recently, an association between autism and microdeletion/microduplication events of 16p11.2 has also been shown.84 85 Copy number variations of various sizes and locations have been reported during the recent years within the ICCA region in normal individuals as well81 86–97 (database of genomic variants at http://projects.tcag.ca/variation/?source = hg18).
CONCLUSION
It is clear that common and possibly unsuspected molecular pathways shared by a subset of epileptic seizures and abnormal movement disorders exist. Genetic studies of the rare families where such brain disorders co-segregate in a Mendelian fashion represent powerful tools for unravelling the molecular basis of these disorders as well as the basis for their close and intertwined relationship. A missense mutation was found in the KCNMA1 gene in one single family with absence epilepsy and PD. Other genes can be involved in pure PD of the non-kinesigenic type (MR1), in BFIS only when associated with hemiplegic migraine (ATP1A2), or in BFNIS, a related albeit different syndrome (SCN2A). However, the major chromosome 16 linked gene that is responsible for the most frequent and familial association of BIS with PD (ICCA syndrome), as well as for most isolated forms of BFIS, remains unknown. The search for the ICCA gene has been hampered by the complicated genomic architecture of the highly duplicated pericentromeric ICCA area. In this regard, future identification of the ICCA gene should provide important insights into the genetic defects and the pathophysiology of BIS, PD, and their tight association in the context of ICCA and ICCA-like syndromes.
Obtained.
Acknowledgments
We thank Professor SL Thein for helpful comments on the manuscript.
REFERENCES
Footnotes
While this review was under editing process, two articles (Suls et al, Brain, Advance Access, published 2008 June 24th, PMID: 18577546; Weber et al, J Clin Invest, June 2008, vol 118, PMID: 18577546) reported on the mutations of the chromosome 1p34.2 SLC2A1 gene, encoding the glucose transporter GLUT1 (OMIM 138140), in families presenting with epilepsy associated with paroxysmal exertion induced dyskinesia. These results add to the complexity of the association of epilepsy with paroxysmal dyskinesia as some of the families reported in these articles also presented with hematological symptoms. They also bring novel insights into one of the possible mechanisms that may cause the association of epilepsy with paroxysmal dyskinesia, and provide a potential treatment option (ketogenic diet) at least for the forms that are linked to SLC2A1 mutations.
Funding: This work was supported in part by a grant from the Picardie county as well as by INSERM and by GIS/Institut des Maladies Rares.
Competing interests: None.