Article Text

Abstract
RAS proteins play key roles in normal cell growth, malignant transformation and learning and memory. Somatic mutations in RAS genes and several of their upstream and downstream molecules result in different human malignancies. In recent years germline mutations in genes coding for components of the RAS signalling cascade have been recognised in a group of phenotypically overlapping disorders, referred to as the neuro-cardio-facial-cutaneous syndromes. These present with variable degrees of psychomotor delay, cardiac abnormalities, facial dysmorphism, short stature, skin defects and increased cancer risk. These findings point to important roles for this evolutionary conserved pathway not only in oncogenesis, but also in cognition, growth and development. Other constitutional disorders caused by mutated RAS pathway genes point to involvement of the RAS-MAPK pathway in immune modulation and vascular development.
Statistics from Altmetric.com
RAS GENES AND CANCER
RAS genes were first identified as homologues of rodent sarcoma virus genes. In 1982 human DNA sequences homologous to the transforming oncogenes of the v-Harvey (HRAS) and Kirsten (KRAS) rat sarcoma virus were identified in DNA sequences derived from a human bladder and a human lung cancer cell line, respectively.1 The findings pointed to the important role of RAS genes as oncogenes and it soon became clear that encoded proteins were constitutively active due to point mutations in the RAS genes.
RAS proteins control signalling pathways that are key regulators of normal cell growth. About 20–30% of all tumours harbour an activating mutation in one of the RAS genes. In these tumours, the activated RAS protein contributes significantly to several aspects of the malignant phenotype, including the deregulation of cell growth, programmed cell death and invasiveness, and the ability for neo-angiogenesis. Yet another important function of RAS and its downstream effectors is their involvement in neuronal processes such as learning, memory and synaptic plasticity.2–6
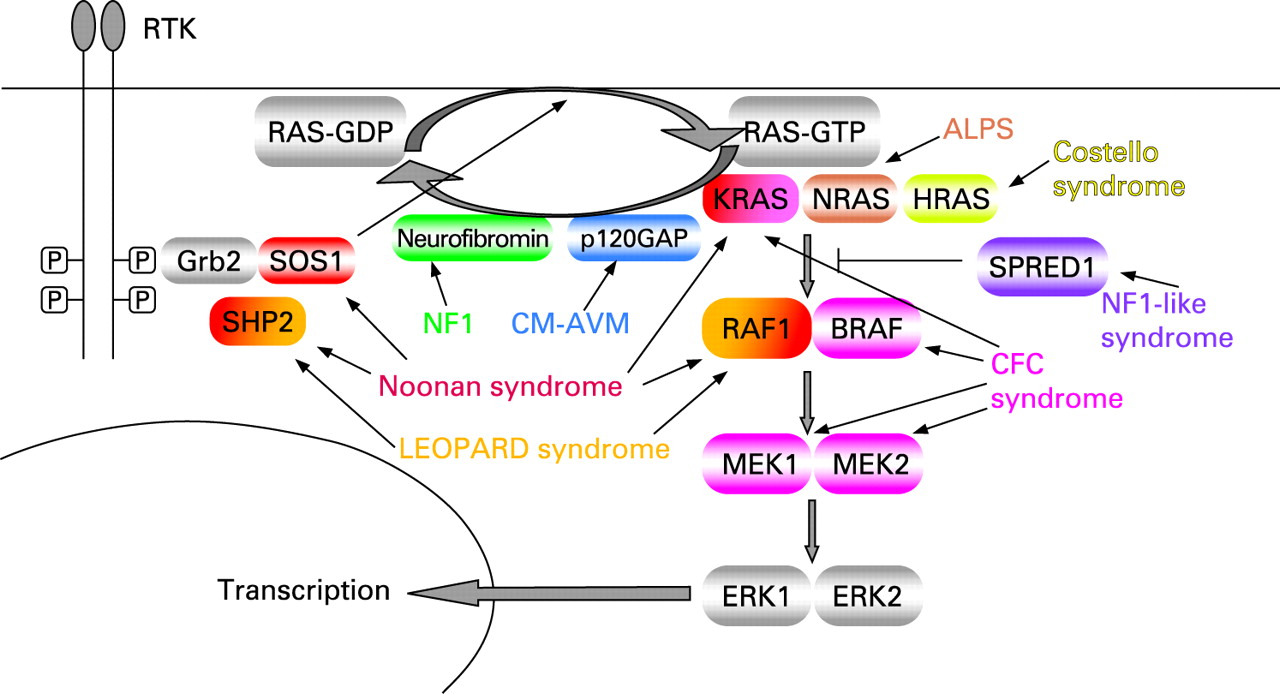
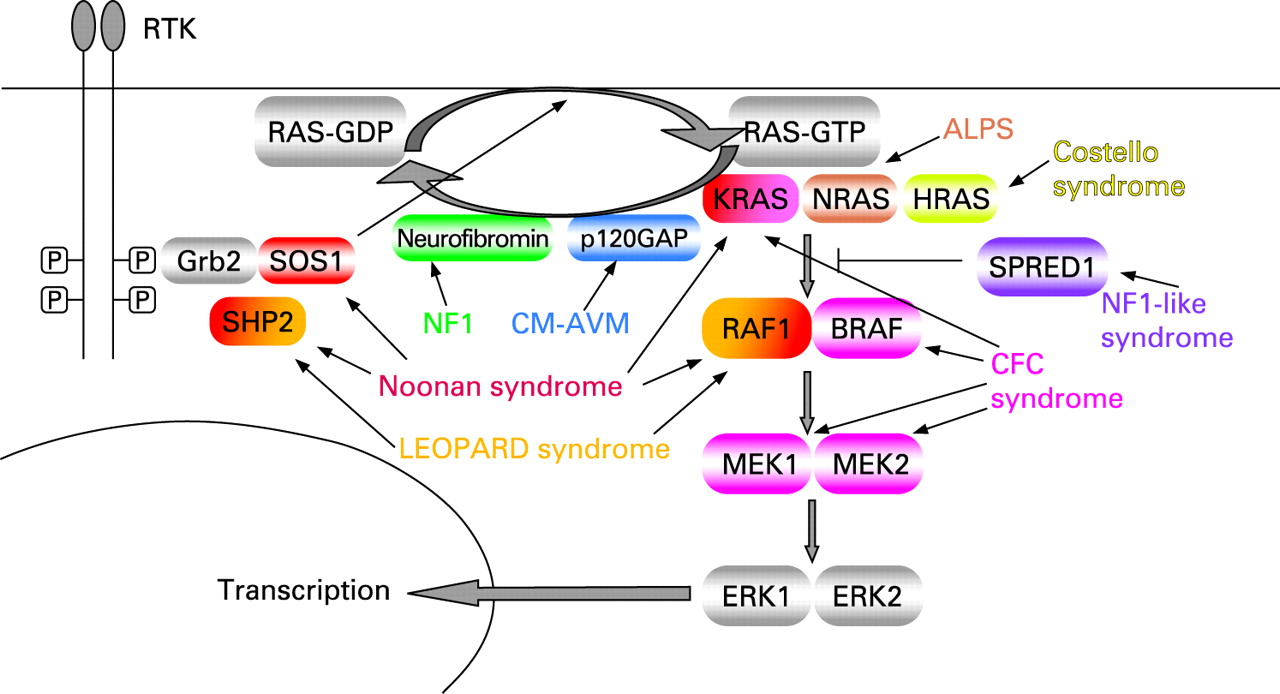
RAS proteins are guanosine nucleotide-bound proteins which cycle between an active GTP-bound and an inactive GDP-bound conformation. They can be activated when a growth factor binds to a receptor tyrosine kinase such as the epidermal growth factor receptor (EGFR). This results in dimerisation and autophosphorylation of the receptor, which binds to the SH2 domain of the adaptor protein GRB2. Through its SH3 domains, GRB2 is bound to SOS, which is thus recruited to the plasma membrane. SOS proteins (SOS1 and SOS2) are important RAS-GEFs (guanosine nucleotide exchange factors) which catalyse exchange of GDP bound to RAS for GTP. The increased proximity of SOS to membrane bound RAS results in increased nucleotide exchange on RAS. Many other receptor types, including the G protein-coupled receptors, can activate RAS through stimulation of exchange factors. RAS-GTP has different effector molecules of which the serine-threonine kinase RAF (MAPKKK = MAPkinasekinasekinase) is the most important. Activated RAF-kinases phosphorylate MEK (MAPKK = MAPkinasekinase), which in turn phosphorylates and activates ERK ( = MAPKinase). ERK/MAPK has many nuclear as well as cytosolic substrates, such as transcription factors and signalling proteins. The result of signalling through this pathway is a change in the pattern of gene expression which may stimulate cell proliferation, promote cell survival or control cell differentiation. Signalling through this cascade is terminated when GTP is hydrolysed to GDP either by the intrinsic GTP-ase activity of RAS (slow), or by GTP-ase activating proteins (GAPs; fast), such as neurofibromin, the protein product of NF1, or p120 GAP, the protein product of RASA1. RAS-MAPK signalling was reviewed recently as well as RAS GDP-GTP cycling.7–9
In addition to the RAF-MAPK pathway, RAS can also activate several other effectors, including type I phosphoinositide 3-kinases (PI3Ks), RALGDS proteins and phospholipase Cϵ (PLCϵ). These pathways will not be discussed in detail here. A novel family of negative regulators of the RAS-MAPK pathway has recently been described. Sprouty proteins were initially identified in a genetic screen as inhibitors of Drosophila FGF receptor signalling during tracheal development10 and were subsequently found to be suppressors of Ras-signalling.11 12 Somewhat later Spred proteins, which contain a Sprouty related domain, were found to modulate Ras–Raf interaction and MAP kinase signalling.13
Mutations in different components of this RAS-MAPK pathway are found in human tumours.
Some 20–30% of human tumours have activating point mutations in RAS, the prevalence being the highest in adenocarcinoma of the pancreas (90%), colon (50%), thyroid (50%), lung (30%), and melanoma (25%).14 Mutations are found most frequently in KRAS (about 85% of total), less in NRAS (about 15%), and rarely in HRAS (<1%). These three RAS family members share 85% amino acid sequence identity. They are widely expressed, with KRAS being expressed in almost all cell types. Knockout studies have shown that Hras and Nras are not required for development in the mouse, whereas Kras is essential.15
Mutational hotspots in RAS genes lead to changes in amino acids G12, G13 and Q61. Mutations in these residues impair the slow intrinsic GTP-hydrolysis of RAS proteins as well as their response to GAPs, which results in constitutive active GTP-bound RAS and constitutive activation of downstream effectors such as the MAPK pathway.16 RAF is the downstream effector of RAS in the MAPK pathway, and is also a proto-oncogene. It has three isoforms of which BRAF is the most effective in activation of the MAPK pathway. About 7% of human cancers harbour BRAF mutations with mutations found in 70% of melanomas, 30% of thyroid cancers, 15% of colon cancers and at lower frequencies in several other cancer types.17 Most of the mutations are located within the kinase domain, the most frequent leading to the amino acid substitution V600E. This mutation results in a constitutive activation of the kinase domain and its downstream targets MEK and ERK.18 Interestingly, it appears that one hit to the MAPK pathway is sufficient for its activation since the current data show very little overlap of BRAF and RAS activating mutations in cancer samples. For example, a recent study in colorectal cancers found 36% BRAF mutations and 18% RAS mutations, yet each mutation occurred exclusively.19
RAS signalling pathways are also active in tumours in which growth factor receptor tyrosine kinases have been overexpressed, such as EGFR and ERBB2 (also known as HER2/neu) in breast, ovarian and stomach carcinomas.20
GERMLINE MUTATIONS IN COMPONENTS OF THE RAS-MAPK PATHWAY
In the past years it has become obvious that constitutional mutations in genes of the evolutionary conserved RAS-MAPK pathway can cause human disorders. It was already known for some time that inactivating mutations in the NF1 gene are responsible for neurofibromatosis type 1 (NF1, Von Recklinghausen’s disease)21–24 and that heterozygous missense mutations in PTPN11, coding for SHP2 which positively modulates RAS signalling, are found in 50% of Noonan25 and in most LEOPARD syndrome patients.26 27 In 2005 de novo heterozygous missense mutations in HRAS were found to be responsible for Costello syndrome.28–31 This was the first time that germline mutations in a member of the oncogenic RAS family were described in a human disorder. Somewhat later mutations in KRAS were discovered in <2%,32–34 and mutations in SOS1 in approximately 20% of PTPN11-negative Noonan syndrome patients.35–37 Mutations in RAF1 (or CRAF), a RAF-isoform not typically mutated in human cancer, are found in Noonan and LEOPARD syndrome patients, especially those presenting with hypertrophic cardiomyopathy.38 39 In CFC syndrome mutations in KRAS, as well as in the downstream effectors BRAF, MEK1 and MEK2 were found.40 41 Finally, germline mutations in SPRED1 have been reported in patients with an NF1-like disorder.42 NF1, LEOPARD, Noonan, Costello and cardiofaciocutaneous (CFC) syndrome are phenotypically overlapping disorders which are referred to as the “neuro-cardio-facial-cutaneous (NCFC) syndromes”.43 These disorders all share a variable degree of mental retardation or learning disabilities, cardiac defects (often pulmonary valve stenosis and hypertrophic cardiomyopathy), facial dysmorphism, short stature, macrocephaly and skin abnormalities. Moreover, an increased risk for malignancy has been described in these patients. The significant phenotypical overlap between these disorders can now be explained by the common deregulation of the same signalling pathway.
Germline mutations in components of the RAS-MAPK pathway have also been found in other conditions. Autoimmune lymphoproliferative syndrome (ALPS) is the most common genetic disorder of lymphocyte apoptosis and is characterised by chronic accumulation of non-malignant lymphocytes, defective lymphocyte apoptosis, and an increased risk for the development of haematological malignancies. Most of the individuals with ALPS have impaired extrinsic Fas-receptor mediated apoptosis due to mutations in the CD95 pathway. In a male patient with an unusual clinical presentation of ALPS, with lifelong overexpansion of lymphocytes and two malignancies (childhood leukaemia and early adulthood lymphoma), a germline NRAS G13D mutation was reported.44 In contrast to germline KRAS and HRAS mutations, this NRAS mutation did not cause any developmental defects. This finding underlines the functional heterogeneity of the different RAS isoforms and the role of NRAS in immune regulation and in haematopoietic cells. These roles are illustrated by mouse models where inactivation of Nras causes subtle immune deficiencies,45 whereas activating Nras mutations cause several haematological malignancies.15
Capillary malformation-arteriovenous malformation (CM-AVM) is a vascular disorder which has been linked to a susceptibility locus on chromosome 5q.46 Subsequently heterozygous inactivating mutations in a positional candidate gene, RASA1, the gene encoding p120-GAP, were identified.47 The hallmark of CM-AVM are atypical capillary malformations (CM). In addition to CM, high flow lesions, either arteriovenous malformations (AVM) or arteriovenous fistulas (AVF), are observed. One individual presented with Parkes Weber syndrome (OMIM 608355) characterised by a cutaneous flush with underlying multiple micro-AVFs, in association with soft tissue and skeletal hypertrophy of the affected limb. Six different mutations in six families were identified. Four families had a frameshift, one a nonsense mutation and one a missense mutation in a highly conserved residue. Of the 39 individuals carrying a RASA1 mutation, four were unaffected. The incomplete penetrance together with the localised nature of the lesions suggests a somatic second hit. Somatic nonsense mutations in the SH2 domain of RASA1 have been demonstrated in basal cell carcinomas.48 In CM-AVM no malignancies have been reported, probably because of residual RASA1 activity or compensation by other GAPs. In mouse models heterozygous loss of Rasa1 did not cause any observable phenotype, whereas homozygous loss causes embryonic lethality with defects in vascular development. In addition murine embryos mosaic for Rasa1-/- cells developed localised vascular defects.49 The clinical and molecular aspects of the different RAS-related disorders are listed in table 1 and an overview of the RAS-MAPK pathway and associated disorders is given in fig 1.
NEUROFIBROMATOSIS TYPE 1
Neurofibromatis type 1 (NF1, OMIM 162200) is an autosomal dominant inherited condition characterised by pigmentation abnormalities of the skin (café-au-lait macules, freckling in the armpits and groins), Lisch nodules in the iris and learning and behavioural problems. There is a predisposition for the development of tumours of the peripheral nerves such as benign neurofibromas and malignant peripheral nerve sheath tumours (MPNST). Rhabdomyosarcoma and neuroblastoma occur at an increased frequency in children with NF1, and in adults with NF1 gastrointestinal stromal tumours and glomus tumours of the fingertips are frequently seen.50 51 In rare situations juvenile myelomonocytic leukaemia (JMML), a myeloproliferative disorder with a severe and often lethal course, is observed. The majority of NF1 patients harbour an intragenic NF1 mutation, but 5% of individuals have a microdeletion of 1.4 megabases in the NF1 region containing at least 11 genes. These patients tend to have a more pronounced tumoural phenotype with an increased risk for development of MPNSTs.52 Moreover these patients are usually taller53 and have more learning problems than individuals with an intragenic NF1 mutation.54 The NF1 protein is 327 kDa with multiple functional domains. One important domain is the GAP related domain which regulates RAS through activation of the RAS GTP-ase. NF1 mutations disturb this GAP activity of the protein resulting in more active RAS and increased signalling through the RAS-MAPK pathway. The NF1 gene acts as a tumour suppressor gene22 51 55 and in NF1 related tumours the normal copy of the NF1 gene is inactivated. Somatic NRAS or KRAS mutations are responsible for approximately 25% of sporadic cases of JMML,56 while in NF1 patients a somatic inactivation of the normal NF1 allele is found.57 Some NF1 patients also have features of Noonan syndrome and “neurofibromatosis–Noonan syndrome” has been described as a separate entity, but NF1 gene mutations represent the major molecular event underlying neurofibromatosis–Noonan syndrome.58
COSTELLO SYNDROME
Costello syndrome (OMIM 218040), first described by Costello,59 60 is a sporadic occurring disorder characterised by a high birth weight, neonatal feeding problems with subsequent failure to thrive and postnatal growth retardation, redundant skin of the neck, palms and soles, mental retardation, characteristic coarse facial features, relative macrocephaly, cardiac abnormalities and tumour predisposition. Typical facial features in Costello syndrome are a wide forehead, epicanthal folds, depressed nasal bridge, low set, posteriorly rotated ears with thick lobes, full cheeks and thick lips. The hair is soft, curly and sparsely implanted (fig 2A). Palmar and plantar creases are deep. Cardiac defects are found in 63% of patients and include structural heart malformations, most frequently pulmonary valve stenosis, hypertrophic cardiomyopathy and cardiac rhythm disturbances, especially atrial tachycardia.61 Tumour risk in Costello syndrome is estimated to be 11–17%.31 62 The most common tumour observed is rhabdomyosarcoma, followed by neuroblastoma and bladder carcinoma.63 Benign papillomata in the peri-oral and peri-anal region are a distinct feature. Mental retardation is mild to moderate with the IQ ranging from 25–85.64

{kind=link}
{kind=link}
In 2006 Aoki used a candidate gene approach to identify de novo heterozygous missense mutations in the HRAS gene in individuals with Costello syndrome.28 This study was the first report of germline RAS mutations as a cause of human disease. Surprisingly, these HRAS germline mutations affect the same amino acid residues (12 and 13) that are mutated in cancer. Later reports confirmed the presence of HRAS mutations in 85–90% of Costello syndrome patients, with G12S being the most prevalent substitution and G12A the second most prevalent.29–31 One report described somatic mosaicism for an HRAS codon 12 mutation.65 As mentioned, these mutations derange RAS GTP-ase activity and cause constitutive activation of the RAS-MAPK pathway. Mutations leading to substitutions in other HRAS amino acids have been reported in some patients with Costello syndrome: K117R31 66 and A146T.67 The K117R mutation has similar activating potential as the classical codon 12 mutations, although the recombinant protein demonstrates normal intrinsic GTP hydrolysis and responsiveness to GAPs. Because of an increased nucleotide dissociation and reassociation rate resulting from a perturbation of guanine nucleotide binding, and because of a physiologically high intracellular concentration of GTP, this mutant RAS protein is predicted to accumulate in the active GTP-bound conformation in vivo.66
NOONAN SYNDROME
Noonan syndrome (OMIM 163950) is an autosomal dominant inherited disorder with an estimated incidence of 1/1000 to 1/2500 live births. Patients have a short stature and specific facial features such as hypertelorism, ptosis, epicanthal folds, low implanted and posteriorly rotated ears and clear blue irises. The neck is broad and often webbed with a low posterior hairline. Thoracic abnormalities including widely spaced nipples and a superior pectus carinatum/inferior pectus excavatum are often present. Congenital heart defects are found in 50–80% with most frequent defects being pulmonic stenosis (20–50%) and hypertrophic cardiomyopathy (20–30%), but also atrial septal defect (ASD), ventricular septal defect (VSD) and tetralogy of Fallot can be found. Less frequent findings are cryptorchidism in boys and bleeding diathesis. Developmental delay is present in 15–35% of patients and is rather mild. Patients are at risk for development of haematological malignancies like JMML as well as solid tumours like rhabdomyosarcoma and giant cell tumours, although the absolute risk is relatively low.
Noonan syndrome had been mapped to a locus on chromosome 12q24.1 by linkage analysis studies68–70 and subsequently germline mutations in the PTPN11 gene have been found in approximately 50% of Noonan syndrome patients.25 PTPN11 is a non-receptor protein tyrosine phosphatase encoding the SHP-2 protein which relays signals from activated tyrosine kinase receptor complexes to downstream signalling molecules, like RAS. PTPN11 mutations occurring in Noonan syndrome are almost always missense mutations and most are gain-of-function mutations, which disrupt an auto-inhibitory interaction between the N-terminal src homology 2 (SH2) domain and the catalytic phosphatase domain. This results in enhanced phosphatase activity and activation of the RAS-MAPK pathway.71 Specific germline PTPN11 mutations are found in Noonan syndrome patients with JMML and in about 35% of sporadic JMMLs, somatic PTPN11 mutations exhibiting stronger activation of SHP-2 were found.72 73
PTPN11 mutations are significantly associated with the presence of pulmonic stenosis, short stature, easy bruising and thorax deformities. PTPN11-negative patients more frequently show cardiomyopathy and less typical facial features.74 The search for additional Noonan associated genes has been hampered for some time by the fact that PTPN11-negative cases are often sporadic.
Finally, in 2006 a germline KRAS mutation was found in a Noonan syndrome patient who presented with a severe clinical phenotype and a JMML-like myeloproliferative disorder.33 Subsequently KRAS mutations were identified in less than 2% of Noonan syndrome patients.32–34 The phenotype associated with KRAS mutations has a broad range varying from mild to severe Noonan syndrome with features of CFC and Costello syndrome. Germline KRAS mutations deregulate RAS-MAPK signalling via two distinct pathogenic mechanisms.75 Decreased GTP-ase activity, both basally and after stimulation with GAPs, a mechanism similar to the one seen in oncogenic mutations, has been reported for Noonan associated KRAS mutations V14I and T58I.33 Another mechanism has been put forward for the germline KRAS mutations V152A and D153V.32 Similar to the HRAS K117R mutation these mutations affect parts of the protein that contribute to the interaction of KRAS with the guanosine nucleotide ring and result in increased GTP/GDP dissociation. This causes accumulation of active RAS, because of the physiological abundancy of GTP in the cell.
In contrast to HRAS mutations, in Costello syndrome overlap between germline KRAS mutations in Noonan syndrome and somatic oncogenic mutations is seldom observed. In functional assays Noonan associated mutations have less pronounced stimulating effects than known oncogenic mutations and intrinsic GTP-ase activity is intermediate between wild-type KRAS and oncogenic G12D KRAS.33 One hypothesis to explain these findings is that KRAS has important roles during embryogenesis and strongly activating KRAS mutations, like the ones found in cancer, would not be tolerated in the germline and would result in embryonic lethality.76 This idea is reflected in the fact that Kras knockout mice die in utero, whereas Hras or Nras knockouts do not.15
Heterozygous missense mutations in the guanine exchange factor SOS1 were also identified in 17–28% of Noonan syndrome patients.35–37 Phenotypically these patients often present with pulmonary valve disease, while ASD is less frequent than in patients with PTPN11 mutations. Ectodermal abnormalities, including facial keratosis pilaris, sparse eyebrows and curly hair, are frequently observed and are similar to the skin features found in CFC syndrome (fig 2B). However, in contrast to CFC syndrome, mental retardation is less frequent in patients with SOS1 mutations. A high prevalence of normal growth was observed in the initial studies,35 36 but this could not be confirmed in a later study.37 Noonan syndrome associated SOS1 mutations cluster at amino acid residues implicated in the maintenance of SOS1 in its auto-inhibited form. Release of autoinhibition as a result of mutations enhances RAS and ERK activation in vitro. A SOS1 frameshift mutation of unknown biochemical consequence has been reported in hereditary gingival fibromatosis.77 Until recently the occurrence of SOS1 mutations in cancer had not been evaluated. A recent study found only three missense changes in SOS1 in a series of 810 primary malignancies and acute myelogenous leukaemias as well as several neuroblastoma cell lines.78 The authors concluded that SOS1 is not a major human oncogene in most cancers and that Noonan syndrome patients with SOS1 mutations may not be at risk of developing cancer.
In 2007 heterozygous missense mutations in RAF1 were reported in a subset (3–17%) of Noonan and LEOPARD syndrome patients.38 39 Whereas hypertrophic cardiomyopathy is rare in Noonan syndrome caused by PTPN11 or SOS1 mutations, it is frequently observed in association with RAF1 mutations (76% prevalence of hypertrophic cardiomyopathy in comparison with 18% in the general Noonan population).38 In addition to the specific cardiac phenotype hyperpigmented cutaneous lesions like multiple naevi, lentigines or café-au-lait spots were found in one third of individuals with RAF1 mutations (fig 2C). Mutations cluster in the CR2 domain in exon 7 (around Ser 259) and in the CR3 domain in exons 14 and 17 (around Ser 612). Hypertrophic cardiomyopathy is associated with the mutation clusters in exon 7 and 17. The inactive conformation of the RAF1 protein (with interaction between the N-terminal and C-terminal region) is stabilised by 14-3-3 protein dimers that bind to phosphorylated Ser 259 and Ser 621. Thus mutations at these sites could alter RAF1s activation status and kinase activity, although the exact mechanism is not clear. These mutations also exhibit the highest kinase activity and MAPK activation in functional assays. The majority of the Noonan related RAF1 mutations differ from the alterations that are observed in cancer. Missense changes in RAF1 are observed though very rarely in cancer and can occur somatically79 or in the germline.80 The importance of RAF1 in cardiac development is confirmed in animal studies. Zebrafish embryos with morpholino knockdown of raf1 show an enlarged, unbent heart tube39 and cardiac specific loss of raf1 in mice results in dilated cardiomyopathy.81
LEOPARD SYNDROME
LEOPARD syndrome (OMIM 151100) is an acronym which refers to the combination of multiple Lentigines, ECG abnormalities, Ocular hypertelorism, Pulmonary valve stenosis, Abnormalities of the genitalia, Retardation of growth, and sensorineural Deafness. It has an autosomal dominant inheritance mode. The most typical features are multiple lentigines, a “Noonan-like” appearance and congenital cardiac abnormalities (fig 2D). Apart from pulmonic stenosis, subvalvular aortic stenosis and progressive hypertrophic cardiomyopathy are also found. Mental retardation is usually mild, if present. Specific heterozygous missense mutations in PTPN11 are found in these patients.26 27 Biochemical in vitro studies point to a dominant negative mechanism for LEOPARD associated mutations, resulting in diminished SHP-2 catalytic activity in contrast to the enhanced SHP-2 activity found for Noonan syndrome and JMML associated PTPN11 mutations.71 The question remains how gain- and loss-of-function SHP-2 mutants can cause phenotypically similar disorders. Moreover, gain-of-function RAF1 mutations have been reported in LEOPARD syndrome cases as well38; thus the pathogenesis of LEOPARD syndrome cannot simply be understood by reduced RAS-MAPK signalling in one situation and increased signalling in the other situation.
CARDIOFACIOCUTANEOUS SYNDROME
CFC syndrome (OMIM 115150) is a rare sporadic occurring disorder, which has been considered in the past as a severe form of Noonan syndrome. PTPN11 mutations were not found in patients with typical CFC syndrome, pointing to the distinct aetiology of the syndrome.82 Affected individuals often present with polyhydramnios during pregnancy and postnatal failure to thrive. Typical facial findings are a high forehead with bitemporal constriction, downslanting palpebral fissures, a depressed nasal bridge, and posteriorly rotated ears with prominent helices. Differential diagnosis with other NCFC syndromes is mainly based on the typical ectodermal abnormalities: dry, hyperkeratotic, scaly skin and sparse, curly and friable scalp hair. Ulerythaema ophryogenes (absent eyebrows with hyperkeratosis) is a distinct feature. At an older age palmoplantar hyperkeratosis and lymphoedema may be observed (fig 2E). Psychomotor delay in these patients is moderate to severe. One or more cardiac abnormalities are found in 75% of individuals, with pulmonic stenosis in 45%, ASD in 22%, and some form of myocardial disease (most often hypertrophic cardiomyopathy) in 40%.83 BRAF mutations are found in 40–78% of CFC patients.40 41 Two clusters of mutations were identified, one (with the most prevalent mutation Q257R) in exon 6 in the cysteine-rich domain, and the other in the protein kinase domain (mutations in exons 11, 12, 14 and 15). These rarely overlap with cancer associated BRAF mutations.
In functional assays increased as well as decreased kinase activity was found resulting in respectively increased and decreased MAPK activation and ELK transactivation. The same observation of possible decreased kinase activity has been made for oncogenic BRAF mutations.18 84 However, these oncogenic kinase impaired mutants still activated ERK in vivo through a mechanism activating RAF1 (CRAF).84 Mutations in the downstream effectors MEK1 and MEK2 have each been found in approximately 5–10% of CFC individuals.41 85–87 These mutant MEK proteins increased levels of phosphorylated ERK in vitro.41 In some individuals with a clinical diagnosis of CFC syndrome KRAS mutations have been reported.33 40 While some studies found no significant differences in phenotypical features between patients with BRAF, MEK1/2 or KRAS mutations,85 other studies did suggest differences. A lower incidence of skin abnormalities including ichthyosis, hyperkeratosis and haemangioma was observed in KRAS-positive individuals,40 and MEK mutations have been reported in association with a less severe phenotype compatible with normal mental development86 and with slightly different craniofacial features.87
In contrast to the other NCFC syndromes, CFC syndrome is believed to have no increased malignancy risk. However, acute lymphoblastoid leukaemia (ALL) has been observed in two patients with CFC syndrome and a germline BRAF mutation,40 88 and a metastatic hepatoblastoma has been reported in another patient with an MEK1 mutation.89 Notably, this last patient received immunosuppressive medication after a heart transplantation because of severe hypertrophic cardiomyopathy. Thus possible tumour predisposition should be kept in mind in patients presenting with CFC syndrome. Based on the phenotypical overlap between CFC and Costello syndrome, germline BRAF, MEK and KRAS mutations have been found in patients diagnosed with Costello syndrome.86 90 91 However, polyhydramnios, growth hormone deficiency, cardiac arrhythmias and papillomata are more common in HRAS positive individuals, whereas pulmonic stenosis associated with ASD2 occurs more frequently in BRAF- or MEK-positive individuals.91 Because of the additional marked difference in malignancy risk it has been suggested to reserve the diagnosis of Costello syndrome for those individuals with proven HRAS mutations.86 91 Other authors suggested the classification of all individuals with germline KRAS mutations as Noonan syndrome.92 Patients with mutations in downstream molecules of RAS (BRAF, MEK1/2) would then be classified as having CFC syndrome, while mutations in KRAS and upstream signalling molecules cause Noonan syndrome.
NEUROFIBROMATOSIS TYPE 1 LIKE SYNDROME
Recently a new disorder belonging to the group of NCFC syndromes has been described— neurofibromatosis type 1-like syndrome (OMIM 611431).42 Affected individuals present with an autosomal dominant inherited condition consisting of multiple café-au-lait spots, axillary freckling, macrocephaly and at times a Noonan-like facial appearance (fig 2F). Several patients have learning difficulties. Whether the disorder is associated with an increased cancer risk is not clear at this moment. One individual died of lung cancer and another presented with renal cancer in childhood and a colon adenoma in adulthood. Several adults had multiple benign lipomas. Although the phenotype closely resembles NF1 some typical NF1 features are systematically absent such as Lisch nodules in the iris, neurofibromas and central nervous system tumours. Mutations in the NF1 gene were excluded. Instead heterozygous loss-of-function mutations in the negative RAF regulator SPRED1 were identified. Truncating mutations resulted in increased RAF1-kinase activity and phosphorylated levels of MEK and ERK, as well as increased activity of the downstream transcription factor ELK1. A second hit in SPRED1 was found in melanocytes from a café-au-lait spot of an affected individual indicating that this clinical feature is caused by a biallelic inactivation of the SPRED1 gene as also reported in NF1 associated café-au-lait spots (biallelic NF1 mutation).93
CONCLUSIONS
The importance of the RAS-MAPK signalling pathway in human oncogenesis has been recognised for a long time. The observation of germline mutations in different components of this pathway as the cause of the NCFC syndromes points to its additional importance in cognition, development and postnatal growth. However, there is an important clinical heterogeneity between mutations in the different genes. Germline mutations in NRAS and RASA1 cause disorders of immune regulation and vascular development, respectively, without any developmental defects.
Not all cases of Noonan syndrome are explained by the set of genes described in this review and future research will show if even more genes coding for proteins of the RAS-MAPK pathway are involved.
How mutations in different components of the same signalling pathway result in different yet overlapping phenotypes is not fully understood. A common mechanism seems to be hyperactivation of the pathway. However, some CFC associated BRAF alleles result in decreased in vitro kinase activity, and for LEOPARD associated PTPN11 mutations even a dominant negative effect has been suggested. Phenotypic variation could result from different expression patterns of mutated genes, as well as from different mechanisms of disturbing RAS signalling through specific interactions with effector and regulatory proteins for different mutants. Variability could also result from feedback mechanisms that can affect upstream molecules (like RAS) but not downstream molecules.43 Moreover, RAS and its upstream molecules also affect other pathways than the MAPK pathway.
Other unresolved questions are how oncogenic mutations in some genes can be tolerated in the germline and what defines the differences in malignancy risk between the different syndromes, different genes and even different mutations.
While the recent discoveries create many new questions they also create new hopes for treatment by targeting the RAS-MAPK pathway. Tyrosine kinase inhibitors are already successful in cancer therapy and, for example, the RAF-inhibitor sorafenib has proven its advantage in renal cell and hepatocellular carcinoma. In vitro studies show a sensitivity to MEK inhibition for different mutated proteins found in CFC syndrome.94 In mouse studies the learning problems caused by heterozygous NF1 knockout could be reversed by farnesyltransferase inhibitors and statins that inhibited RAS hyperactivation by reducing the level of isoprenylated RAS.95 The first clinical studies assessing the effect of statins on cognition in NF1 patients are underway.96 If these studies show any effect, then this type of pharmacological treatment might also be studied in the larger group of RAS-MAPK syndromes.
Acknowledgments
ED is predoctoral researcher of the Fonds voor Wetenschappelijk Onderzoek-Vlaanderen.
REFERENCES
Footnotes
Competing interests: None.
Funding: This work is supported by research grants from the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (G.0578.06 and G.O551.08 to EL); the Interuniversity Attraction Poles (IAP) granted by the Federal Office for Scientific, Technical and Cultural Affairs, Belgium (2007–2011; P6/05) (EL) and by a Concerted Action Grant from the K U Leuven (EL).
Patient consent: Parental consent obtained.