Article Text

Abstract
Hirschsprung disease (HSCR, aganglionic megacolon) represents the main genetic cause of functional intestinal obstruction with an incidence of 1/5000 live births. This developmental disorder is a neurocristopathy and is characterised by the absence of the enteric ganglia along a variable length of the intestine. In the last decades, the development of surgical approaches has importantly decreased mortality and morbidity which allowed the emergence of familial cases. Isolated HSCR appears to be a non-Mendelian malformation with low, sex-dependent penetrance, and variable expression according to the length of the aganglionic segment. While all Mendelian modes of inheritance have been described in syndromic HSCR, isolated HSCR stands as a model for genetic disorders with complex patterns of inheritance. The tyrosine kinase receptor RET is the major gene with both rare coding sequence mutations and/or a frequent variant located in an enhancer element predisposing to the disease. Hitherto, 10 genes and five loci have been found to be involved in HSCR development.
Statistics from Altmetric.com
Harald Hirschsprung, a Danish paediatrician, first described in 1888 two unrelated boys who died from chronic severe constipation with abdominal distension resulting in congenital megacolon.1 The absence of intramural ganglion cells of the myenteric and submucosal plexuses (Auerbach and Meissner plexuses, respectively) downstream of the dilated part of the colon was recognised as the cause of the disease in the 1940s.2 This allowed a simple and reliable diagnostic confirmation from rectal suction biopsies using histochemical staining for acetylcholinesterase (AchE).3 In 1948, Swenson and Bill developed a surgical procedure4 and the survival of patients uncovered familial transmission of Hirschsprung disease (HSCR).5 In 1974, Bolande proposed the term neurocristopathy for syndromes or tumours involving the neural crest (NC) cells.6 HSCR resulting from an anomaly of the enteric nervous system (ENS) of NC origin is therefore regarded as a neurocristopathy.6–8
Isolated HSCR appears to be of complex, non-Mendelian inheritance with low, sex-dependent penetrance, variable expression according to the length of the aganglionic segment and suggestive of the involvement of one or more gene(s) with low penetrance.5 9 These parameters must be taken into account for accurate evaluation of recurrence risk in relatives. With a relative risk as high as 200, HSCR appears an excellent model to study common multifactorial diseases. The major susceptibility gene is RET, which is also involved in multiple endocrine neoplasia type 2 (MEN 2) and familial medullary thyroid carcinoma (FMTC). Coding sequence mutations are identified in about 50% and 15% of familial and sporadic HSCR cases, respectively. The far most frequent HSCR predisposing event at the RET locus is a haplotype which comprises an SNP lying in an enhancer element of RET intron 1. The identification of modifier genes is currently underway by using various approaches and an international consortium has been settled in 2004 in order to achieve this goal.
HSCR occurs as an isolated trait in 70% of patients, is associated with a chromosomal abnormality in 12% of the cases, and with additional congenital anomalies in 18% of the cases.10–15 In the latter group of patients, some monogenic syndromes can be recognised. Indeed, thus far, genetic heterogeneity in HSCR has been demonstrated with 10 specific genes involved. The aim of this paper is to update a 6 year old review on clinical and molecular data about isolated and syndromic HSCR.
DEFINITION AND CLASSIFICATION
HSCR is a congenital malformation of the hindgut characterised by the absence of parasympathetic intrinsic ganglion cells in the submucosal and myenteric plexuses.2 It is regarded as the consequence of the premature arrest of the craniocaudal migration of vagal neural crest cells in the hindgut between the fifth and 12th week of gestation to form the enteric nervous system (ENS) and is therefore regarded as a neurocristopathy.6 16 While the internal anal sphincter is the constant inferior limit, patients could be classified as short-segment HSCR (S-HSCR: 80% of cases) when the aganglionic segment does not extend beyond the upper sigmoid, and long-segment HSCR (L-HSCR: 20% of cases) when aganglionosis extends proximal to the sigmoid. Four HSCR variants have been reported: (1) total colonic aganglionosis (TCA, 3–8% of cases)17; (2) total intestinal HSCR when the whole bowel is involved17; (3) ultra-short segment HSCR involving the distal rectum below the pelvic floor and the anus18; (4) suspended HSCR, a controversial condition, where a portion of the colon is aganglionic above a normal distal segment.
CLINICAL FEATURES AND DIAGNOSIS
In most cases, the diagnosis of HSCR is made in the newborn period15 due to intestinal obstruction with the following features: (1) delayed of passage of meconium (>24 h after birth); (2) abdominal distension that is relieved by rectal stimulation or enemas; (3) vomiting; and (4) neonatal enterocolitis. Some patients are diagnosed later in infancy or in adulthood with severe constipation, chronic abdominal distension, vomiting, and failure to thrive.19 Finally, although a rare presentation, unexplained perforation of the caecum or appendix should make the diagnosis considered.
On abdominal x ray a distended small bowel and proximal colon, with absence of rectal gas, are common findings. The classical image is a dilated proximal colon with the aganglionic cone narrowing towards the distal gut. On barium enema a small rectum with uncoordinated contractions is seen. The transitional zone represents the site where the narrow aganglionic bowel joins the dilated ganglionic bowel. On a delayed plain x ray taken 24 h after the enema, barium retention is observed. Anorectal manometry shows absence of relaxation of the internal sphincter (rectal inhibitory reflex) in response to rectal distension.20 The reliability of this test becomes excellent from day 12 after birth where the normal rectoenteric reflex is present.21 Suction rectal biopsy remains the gold standard for confirming the diagnosis in most cases demonstrating an increased acetyl cholinesterase activity.22 Nonetheless, full thickness rectal biopsy is the golden standard in reaching the diagnosis. Furthermore, seromuscular biopsies will be needed at operation to define the proximal limit of the aganglionic segment.
DIFFERENTIAL DIAGNOSES
Other causes of intestinal obstruction should be discussed when abdominal distension and failure to pass meconium occur in a newborn infant: (1) meconium ileus resulting from cystic fibrosis; (2) intestinal malformations such as lower ileal and colonic atresia, isolated or occasionally associated with HSCR, intestinal malrotation or duplication; (3) ENS anomalies grouped as chronic intestinal pseudo-obstruction syndromes; and (4) functional intestinal obstruction resulting from maternal infection, maternal intoxication or congenital hypothyroidism.
TREATMENT AND PROGNOSIS
The treatment of HSCR is surgical. After careful preoperative management, the underlying principle is to place the normal bowel at the anus and to release the tonic contraction of the internal anal sphincter. Since the initial protocol of Swenson described in 1948,4 a series of operative approaches, such as the Soave and Duhamel procedures, have been developed.23 24 A one stage procedure is possible when diagnosis is made early, before colonic dilatation, in short segment disease. Otherwise, a primary colostomy is required. For long segment disease and total colonic aganglionosis, temporary enterostomy is often the first step in management before definitive surgery. Laparoscopic and transanal pull-through techniques have been proposed more recently in HSCR surgery.25 These techniques can provide patients with almost scarless surgery. Comparative long term results are pending.26 27 Neuronal precursor cells isolated from the developing human ENS may open the route to cell therapy.28 29 Fistula or stenosis of the anastomosis and enterocolitis are the main short term complications.30 Long term complications include chronic constipation (10–15%) and soiling.31 32 Mortality has been below 6% since the 1980s and may be related to short term complications or caused by the associated malformations.31 However, the treatment of children with TCA remains hazardous.33 34
EPIDEMIOLOGY
The incidence of HSCR is estimated at 1/5000 live births.5 However, the incidence varies significantly among ethnic groups (1.0, 1.5, 2.1, and 2.8 per 10 000 live births in Hispanics, Caucasian-Americans, African-Americans, and Asians, respectively).15 S-HSCR is far more frequent than L-HSCR (80% and 20%, respectively).10 12 There is a sex bias with a preponderance of affected males and a sex ratio of 4/1.35 Interestingly, the male:female ratio is significantly higher for S-HSCR (4.2–4.4) than for L-HSCR (1.2–1.9) (table 1).15 35
MOLECULAR GENETICS IN ISOLATED HSCR
Several genes have been implicated in isolated HSCR, the two major ones being RET and EDNRB.
The RET signalling pathway
The first susceptibility locus was mapped to 10q11.2 in multigenerational families segregating HSCR as an incompletely penetrant autosomal dominant trait.36 37 This region had been targeted because of the observation of an interstitial deletion of chromosome 10q11 in patients with TCA and mental retardation.38 The proto-oncogene RET (REarranged during Transfection), identified as disease causing in MEN 239 40 and mapping in 10q11.2, was regarded as a candidate gene owing to: (1) co-occurrence of MEN 2A and HSCR in some families; and (2) expression in neural-crest derived cells. Consequently, RET gene mutations were identified in HSCR patients (fig 1).41 42 Over 100 mutations have been identified including large deletions encompassing the RET gene, microdeletions and insertions, nonsense, missense and splicing mutations.43–46 There is no mutational hot spot at variance with MEN 2A, where mutations occur in a cluster of six cysteines (exon 10: residues 609, 611, 618, 620; exon 11: residues 630,634),39 40 47 and MEN 2B where the mutation is almost unique (M918T, exon 16, tyrosine kinase domain).48–51 In vitro, MEN 2 mutations have been shown to be activating mutations leading to constitutive dimerisation of the receptor and to transformation,52 while haploinsufficiency is the most likely mechanism for HSCR mutations.53–57 Biochemical studies demonstrated variable consequences of some HSCR mutations (misfolding, failure to transport the protein to the cell surface, abolished biological activity).54 56 58 However, a simple activating versus inactivating model of gene action is not sufficient to explain the co-occurrence of HSCR and MEN 2A in patients with a MEN 2A RET gene mutation.51 59
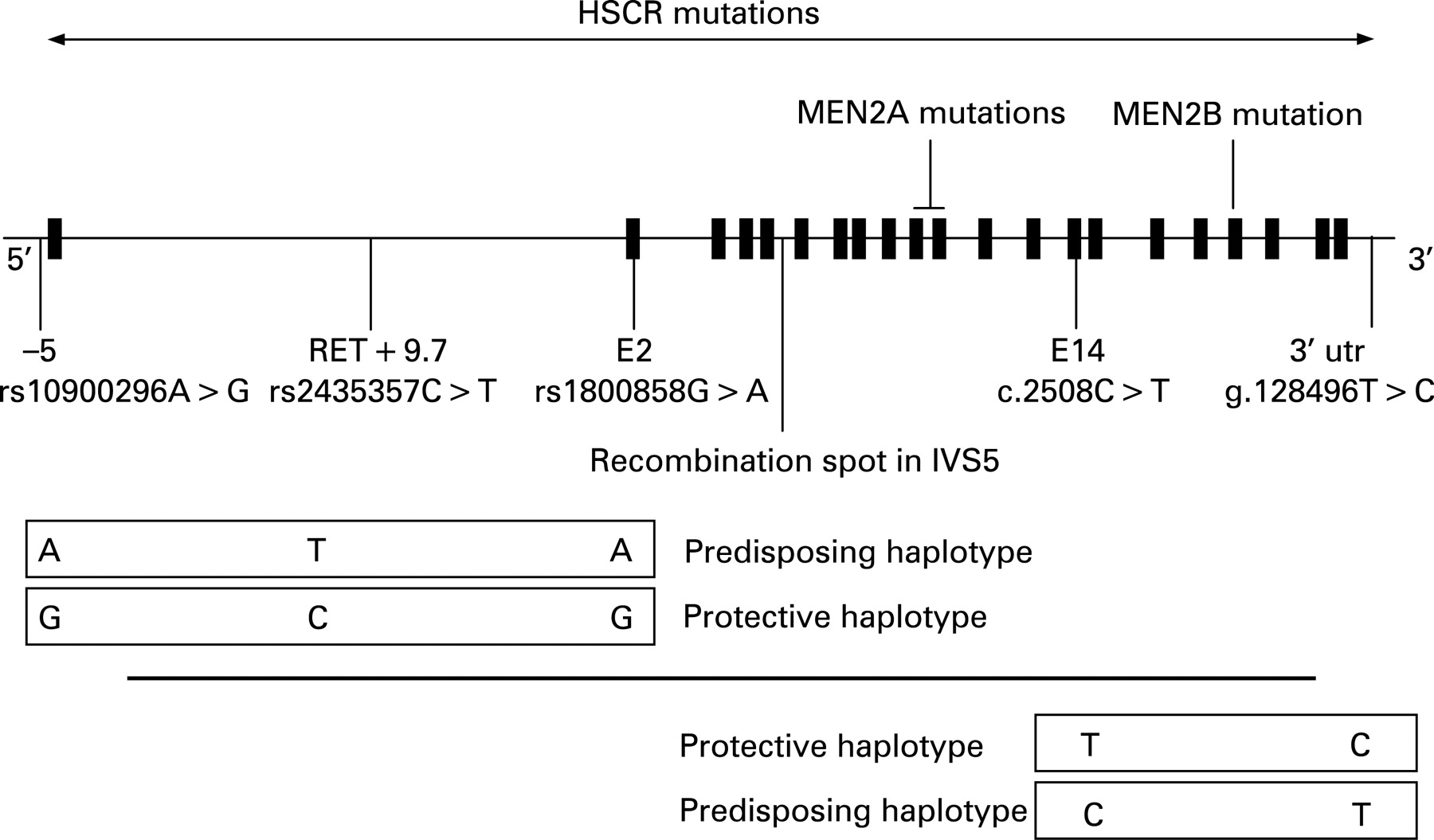
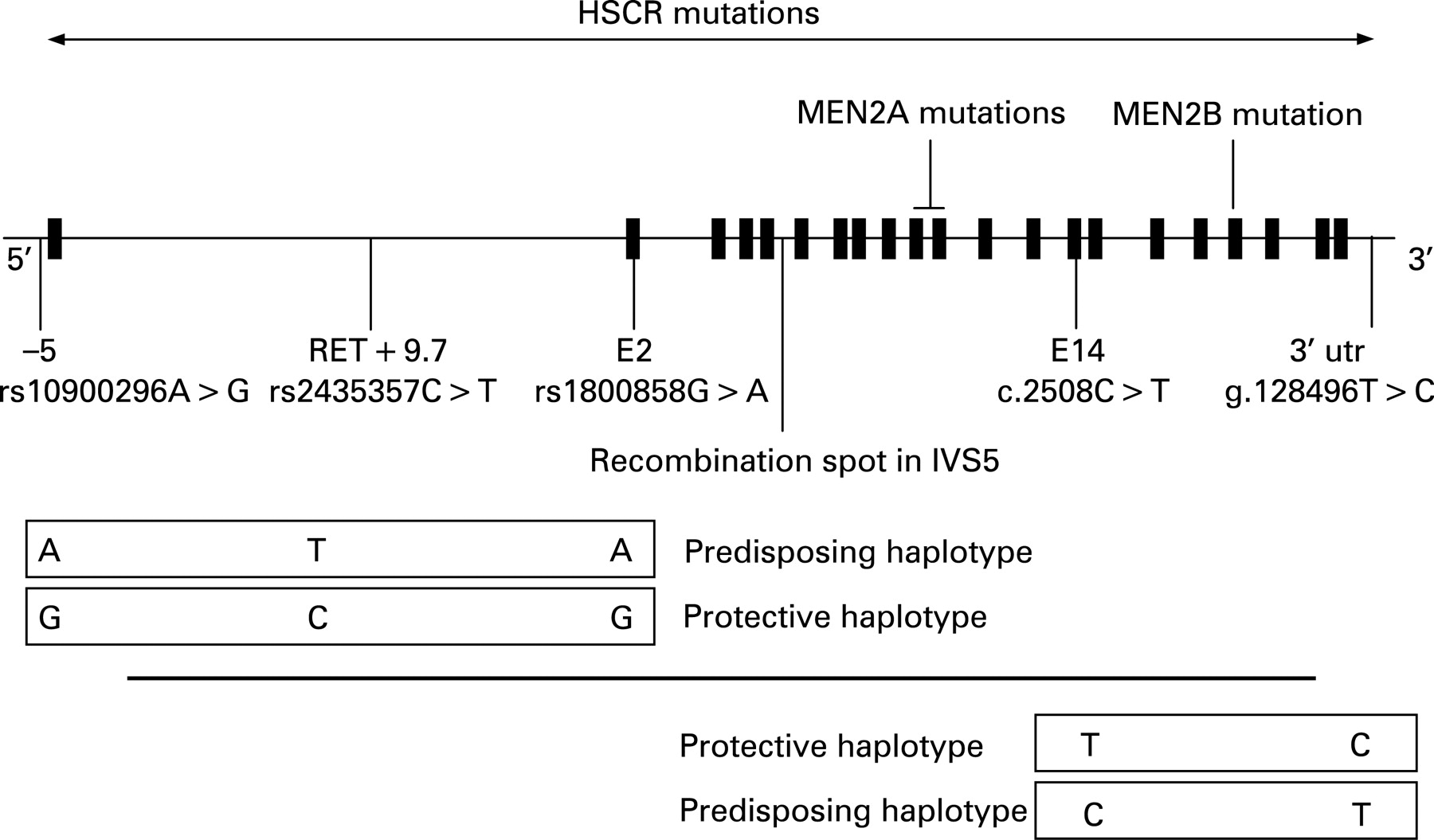{kind=link}
Despite extensive mutation screening, a RET mutation is identified in only 50% of familial and 15–20% of sporadic HSCR cases.43 44 60 61 However, most families with few exceptions are compatible with linkage at the RET locus.62 Case–control and transmission disequilibrium test in several ethnic backgrounds had first pointed to a frequent SNP lying in exon 2 and leading to a silent change as over represented and transmitted in patients (fig 1).63–68 Later, the same observation was made for haplotypes comprising this SNP lying in exon 2 and an SNP at–5 from the transcription start site of RET.69–72 As there was no convincing evidence for a functional role of these two SNPs,71 73 74 the most likely hypothesis was that an ancient, low-penetrant founder locus was in linkage disequilibrium with the haplotype of the two SNPs previously identified and distant of about 25 kb.70 Comparative genomics focused on conserved non-coding sequences and an SNP lying in intron 1 was shown associated to HSCR susceptibility, making a 20-fold greater contribution to risk than coding sequence mutations.75 This T>C SNP lies in an enhancer-like sequence and the T allele reduces in vitro enhancer activity.75 Moreover, this sequence drives reporter expression in tissue consistent with the one of Ret during mouse and zebra fish development.75 76 Interestingly, the frequency of the predisposing T allele varies according to HSCR prevalence in various ethnic backgrounds from about 20–50% in European and Chinese, respectively.75 77 The T allele high frequency in control populations emphasises, as speculated by the oligogenic model, the pivotal role of the RET gene in HSCR susceptibility despite low penetrance. Finally, the penetrance of the T allele for the HSCR trait is both dose-dependent and greater in males than in females.75 Conversely, an SNP lying in the 3′ UTR of the RET gene and lowering stability to RET mRNA degradation has been shown to be under transmitted in HSCR cases.78 Again, this SNP lies on a haplotype that is of variable frequency according to ethnicity (about 8–4% in Caucasian and absent in Chinese).71 79 80 A recombination spot lies on intron 5 at the RET locus.59 66
RET is a 1114 amino acid transmembrane receptor with a cadherin-like extracellular domain, a cysteine-rich region and a intracellular tyrosine kinase domain.81 The role of Ret in mice development has been expanded to kidney,82–84 spermatogenesis85–88 and Peyer’s patch.89 90 Between the two RET major isoforms (RET9 and RET51) with different C-terminal tails as the result of alternative splicing, RET9 is critical for both kidney and ENS development.91
GDNF, known as a major survival factor for many types of neurons, was shown to be the RET ligand by both phenotypic similarities between Ret −/− and Gdnf −/− knock-out mice,92–94 and xenopus embryo bioassays.95 GDNF is a TGF-B related 211 residue protein, proteolytically cleaved to a 134 residue mature protein that homodimerise. To activate RET, GDNF needs the presence of a glycosylphosphatidylinositol (GPI)-linked co-receptor GFRA1.96 97 Four related GPI-linked co-receptors, GFRA1-4,98 and four related soluble growth factor ligands of RET have been identified, namely: GDNF, NTN,99 persephin (PSPN)100 and artemin (ARTN).101 Specific combinations of these proteins are necessary for the development and maintenance of both central and peripheral neurons, and all can signal through RET. GDNF mutations have been identified in only six HSCR patients to date, and could be regarded as a rare cause of HSCR (<5%).102–104 Moreover, GDNF mutations may not be sufficient to lead to HSCR since 4/6 patients have additional contributory factors, such as RET mutations or trisomy 21.102 103 Similarly, an NTN mutation has been identified in one family, in conjunction with a RET mutation.105 Finally, although Gfra1 homozygous knock-out mice are phenotypically very similar to Ret and Gdnf −/− mice, no GFRA1 mutations have been identified in HSCR patients except a deletion at the locus with incomplete penetrance in one family.69 106–109 Worth noting, RET exerts a pro-apoptotic effect that is inhibited by GDNF and some RET gene mutations may impair the control of this activity by GDNF.110
The endothelin signalling pathway
The endothelin pathway was first studied for its vasoconstrictive effect and putative role in hypertension. EDNRB and EDNRA are G-protein-coupled heptahelical receptors that transduce signals through the endothelins (EDN1, 2, 3).111 112
A susceptibility locus for HSCR in 13q22 was pointed out for three main reasons: (1) a significant lod-score at 13q22 in a large inbred Old Order Mennonite community with multiple cases of HSCR113–115; (2) de novo interstitial deletion of 13q22 in several patients with HSCR116; (3) synteny between the murine locus for piebald-lethal (sl), a model of aganglionosis, and 13q22 in human. The critical role of the endothelin pathway in HSCR was demonstrated with the finding that piebald-lethal was allelic to the Ednrb knock-out mouse and harboured an Ednrb mutation (table 2).117 Subsequently, an EDNRB missense mutation was identified in the Mennonite kindred (W276C).118 However, the W276C mutation was neither necessary (affected wild-type homozygotes) nor sufficient (non-affected mutant homozygotes) to cause HSCR, and penetrance was sex-dependent (greater in males than in females).118 piebald-lethal was considered a mouse model for WS4 in humans, and some of the Mennonite affected individuals had pigmentary anomalies and sensorineural deafness in addition to HSCR.113 114 This prompted a screen of the EDNRB gene in WS4, and homozygous mutations in a fraction of WS4 families were found.44 At the same time, an Edn3 mutation was identified in the lethal spotting (sl) natural mouse model for WS4119 and, subsequently, EDN3 homozygote mutations were identified in WS4 in humans (table 2).120 121
Both EDNRB and EDN3 were screened in large series of isolated HSCR patients. While EDN3 mutations were seldom found,130 EDNRB mutations were identified in approximately 5% of the patients.126–129 It is worth mentioning that the penetrance of EDN3 and EDNRB heterozygous mutations is incomplete in those HSCR patients, de novo mutations have not hitherto been observed, and that S-HSCR is largely predominant. Interstitial 13q22 deletions encompassing the EDNRB gene in HSCR patients makes haploinsufficiency the most likely mechanism for HSCR (table 3). Although EDNRB binds all three endothelins, the similarity of phenotype of the Ednrb knock-out mice to that of the Edn3 knock-out mice suggests that EDNRB’s major ligand is EDN3.
Preproendothelins are proteolytically cleaved by two related membrane-bound metalloproteases to give rise to the mature 21-residue endothelin. Ece1 processes only Edn1 and Edn3. Ece1 knock-out mice show craniofacial defects and cardiac abnormalities in addition to colonic aganglionosis.132 A heterozygous ECE1 mutation has been identified in a single patient combining HSCR, craniofacial and cardiac defects (R742C).131
SOX10
The last known mouse model for WS4 in human is dominant megalon (Dom), homozygous Dom mutation being embryonic lethal.152 The Dom gene is Sox10, a member of the SRY (sex determining factor)-like, high mobility group (HMG) DNA binding proteins.125 Subsequently, truncating heterozygote SOX10 mutations have been identified in patients with WS4,122–124 Yemenite deaf-blind-hypopigmentation syndrome153 and WS2 (Bondurand et al in Am J Hum Genet website) but also in patients presenting in addition neurological impairment due to central and peripheral dysmyelination.67 123 The latter combination is known as PCWH for Peripheral demyelination-Central dysmyelinating leucodystrophy-Waardenburg syndrome and Hirschsprung disease. Genotype–phenotype correlation relies on nonsense-mediated decay being effective (WS4) or not (PCWH).154 The penetrance of the HSCR trait appears to be high, although sibs sharing a mutation and discordant for HSCR have been described in one family.124 Therefore, SOX10 is unlikely to be a major gene in isolated HSCR.
Interaction between pathways
Ret and Ednrb signalling pathways were considered biochemically independent. However, G-protein-coupled receptors and tyrosine kinase receptors could be engaged in crosstalk. Moreover an HSCR patient heterozygote for weak hypomorphic mutations in both RET and EDNRB has recently been reported.155 Each mutation was inherited from a healthy parent. Genetic interactions between EDNRB and RET have been demonstrated in the Mennonite population where HSCR predisposition is high (incidence of 1/500).118 156 Finally, no complementation of aganglionosis could be observed in mouse inter crosses between hypomorphic piebald alleles of Ednrb (Ednrbs/s) and a null allele of Ret.156
Sox10 is involved in cell lineage determination and is capable of transactivating MITF synergistically with PAX3.157 Similarly, Ednrb transcripts are either absent or drastically reduced in Dom−/− and Dom+/− mice, respectively.158 Sox10;Ednrb and Sox10;Edn3 double mutants have a severe ENS defect with no enteric progenitor cells extending beyond the stomach at all embryonic stages studied.159 Interestingly, genetic interactions for the HSCR trait have been shown between RET and PHOX2B and BBS genes responsible when mutated for CCHS and Bardet-Biedl syndromes, respectively.148 Such correlation was not found between RET and SOX10. Along these lines, a genome-wide screen aimed at localising modifier genes for the aganglionosis of Dom mice did not point to the Ret locus but, among others, a locus encompassing the PHOX2b gene.160
Taking it all together, several general comments can be made: (1) RET is the major gene in HSCR with either CDS mutations or, more frequently, a low penetrant SNP lying in an enhancer element within intron 1; (2) RET mutation penetrance is incomplete and sex-dependant; (3) genotype–phenotype correlation is poor; 4) HSCR is genetically heterogeneous and due to mutations in distinct pathways; (5) some patients with mutations in more than one HSCR susceptibility gene are known (RET+GDNF, RET+NTN, RET+EDNRB); (6) the RET gene plays a role in HSCR penetrance of some but not all syndromic HSCR (see below).
MULTIGENIC INHERITANCE OF ISOLATED HIRSCHSPRUNG DISEASE
As mentioned above, RET plays a key role in HSCR genesis and multiple genes may be required to modulate clinical expression. On the other hand, genetic heterogeneity where mutation in one of several genes is sufficient for phenotypic expression of HSCR has been demonstrated (RET, EDNRB, EDN3, ECE1). Segregation studies in HSCR showed that the recurrence risk in siblings varies from 1.5–33% depending on the gender and the length of the aganglionic segment in the proband, and the gender of the sibling (table 1).5 35 Consequently, HSCR has been assumed to be a sex modified multifactorial disorder, the effect of genes playing a major role as compared to environmental factors (relative risk of 200).
According to the segregation analysis where an autosomal dominant model in L-HSCR and a multifactorial model in S-HSCR were more likely, different approaches have been chosen to test these hypotheses in L-HSCR and S-HSCR independently.
Linkage analysis in 12 HSCR families with three or more affected individuals in two or more generations where L-HSCR is largely predominant.62 All but one family showed linkage to the RET locus. Mutational analysis identified a nonsense or missense mutation at highly conserved residue in six families, a splice mutation in two families and no coding sequence variation in three families. Linkage to a novel locus in 9q31 was identified only in families with no or hypomorphic RET gene mutation. Therefore, a severe RET mutation may lead to phenotypic expression by haploinsufficiency while hypomorphic RET mutations would require the action of other mutations.
A sib-pair analysis in 49 families with S-HSCR probands.161 This studies shows that only three loci on chromosomes 3p21, 10q11 and 19q12 are both necessary and sufficient to explain the incidence and sibling recurrence risk in HSCR. A multiplicative risk across loci with most affected individuals being heterozygotes at all three loci seems the best genetic model. Finally, linkage to 9q31 was confirmed in the sib-pairs with no or hypomorphic RET mutation.
A genome-wide association study was conducted in 43 Mennonite family trios and identified a susceptibility locus on 16q23 in addition to the loci of the two predisposing genes in this population (RET and EDNRB at 10q11.2 and 13q22, respectively).156
Linkage analysis in a multigenerational HSCR family where the RET gene had been previously excluded, showed linkage to 4q31-q32.162
The route to the identification of modifier genes is now based on various approaches. A differential screen for ENS expressed genes was conducted by a 22 000 probe DNA micro array of embryonic Ret+/+ and Ret−/− mice and identified over 300 genes over expressed in Ret+/+ mice.29 These genes are regarded as critical for enteric neurogenesis and therefore potential candidates in HSCR. By synteny, some lie at candidate modifier loci for isolated HSCR. Other approaches undertook by the HSCR Consortium are microarrays of RNAs from microdissection of enteric neurons and glia on the one hand and 500 k SNP genotyping in trios on the other hand.
SYNDROMIC HSCR
HSCR occurs as an isolated trait in 70% of cases. A chromosomal abnormality is associated in 12% of cases, trisomy 21 being by far the most frequent (>90%). Associated congenital anomalies are found in 18% of the HSCR patients. The one occurring at a frequency above that expected by chance include gastrointestinal malformation, cleft palate, polydactyly, cardiac septal defects and craniofacial anomalies.13 14 The higher rate of associated anomalies in familial cases than in isolated cases (39% vs 21%) strongly suggests syndromes with Mendelian inheritance.14 Assessment of all HSCR patients by a trained dysmorphologist should provide a careful evaluation for recognisable syndromes.
Chromosomal anomalies
A large number of chromosomal anomalies have been described in HSCR patients. Free trisomy 21 (Down syndrome) is by far the most frequent, involving 2–10% of ascertained HSCR cases.5 11–15 In these cases, both the unbalanced sex ratio (5.5–10.5:1 male:female) and the predominance of S-HSCR are even greater than in isolated HSCR. Over-expression of gene(s) on chromosome 21 and predisposing to HSCR has been hypothesised and a susceptibility gene mapping to 21q22 postulated in a Mennonite kindred.114 However, these data were not confirmed.156 Hitherto, coding sequence mutations in genes predisposing to HSCR, RET, EDNRB and GDNF, respectively, were found in only three patients with Down syndrome and HSCR.103 163 However, the common HSCR predisposing RET hypomorphic allele is over represented in patients with Down syndrome and HSCR when compared to patients without HSCR.148
Some chromosomal interstitial deletions reported in combination with HSCR, have been important for the identification of HSCR predisposing genes, namely: (1) 10q11.2 interstitial deletion observed in a few patients with L-HSCR or TCA38 146 leading to the mapping and identification of the first HSCR predisposing gene (RET); (2) 13q22.1-32.1 interstitial deletion in patients with S-HSCR leading to the mapping of the second gene (EDNRB)164–166; (3) 2q22-23 interstitial deletion syndrome in patients with a multiple congenital anomaly–mental retardation (MCA-MR) syndrome with HSCR or severe chronic constipation further delineated as Mowat-Wilson syndrome (table 3),133 134 147 leading to the identification of the ZFHX1B gene (previously named SIP1 gene).135
Rarer chromosomal anomalies reported in combination with HSCR are summarised in table 3. DiGeorge syndrome, mosaic trisomy 8, XXY chromosomal constitution, partial duplication of chromosome 2q, tetrasomy 9p, and 20p deletion, have been observed at least once with HSCR. Interestingly, a patient with S-HSCR, postnatal growth retardation, mild developmental delay, dysmorphic facial features and a deletion at 4p12 encompassing the PHOX2B gene has been reported.167
Syndromes and associated anomalies
Both the recognition of known entities and the delineation of novel ones including HSCR as a feature are of importance for disease prognosis, accurate genetic counselling and search for candidate genes. Syndromes reported associated with HSCR are numerous. Some associations are well characterised with a penetrance of HSCR ranging from 5% to >80% (table 2). For rare disorders, whether an association with HSCR observed once is meaningful or occurred by chance alone is not possible to decide. These conditions are summarised in table 4. Both frequent and occasional associations may be of interest for the identification of susceptibility genes to HSCR.
Syndromes frequently associated with HSCR: neurocristopathies
The NC is a transient and multipotent embryonic structure that gives rise to neuronal, endocrine and paraendocrine, craniofacial, conotruncal heart and pigmentary tissues.7 Neurocristopathies encompass tumours, malformations and single or multifocal anomalies of tissues mentioned above with various combinations. MEN 2, neuroblastoma (NB) conotroncal heart defects and Waardenburg syndromes illustrate each of these categories, and are associated with HSCR.
Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma
Familial medullary thyroid carcinoma (FMTC), MEN type 2A (MEN 2A) and type 2B (MEN 2B) are cancer predisposition syndromes with an autosomal dominant mode of inheritance. MEN 2A is defined by an age-related predisposition to medullary thyroid carcinoma (MTC, 70% by the age of 70 years), pheochromocytoma (50% of cases) and hyperplasia of the parathyroid glands (15–35%). In addition to MTC and pheochromocytoma, individuals with MEN 2B present with oral neuromas, marfanoid habitus and hyperganglionosis of the hindgut.208 Germline missense mutations of the RET gene have been identified in MEN 2A, MEN 2B and FMTC. Both FMTC and MEN 2A can be associated with HSCR in some families.47 175–181 Interestingly, these families present a germline RET mutation of the MEN 2A or FMTC type (see below).47 176–181 This raises the question of whether all individuals with HSCR, regardless of non-contributive family history, should be screened for RET exon 10 and 11 mutations to rule out cancer predisposition (3/160 cases in our series with C609W, C611R and C620R RET gene mutations, respectively).
Neuroblastoma
Neuroblastoma (NB) is the most frequent solid tumour in childhood with an incidence of 1/10 000. The tumour can arise at any site of the sympathetic chain or the adrenal medulla (both originating from NCC). In some families, tumour predisposition segregates through generations with incomplete penetrance.209–211 NB is found associated to HSCR and congenital central hypoventilation (CCHS, see below) in various combinations and, in each combination, heterozygous mutations of the paired-like homeobox 2B gene (PHOX2B) have been identified.137 212–217 However, germeline PHOX2B mutations remain rare in sporadic, isolated NB.215 216
Congenital central hypoventilation syndrome (CCHS, MIM 209880).
Initially termed Ondine’s curse, CCHS is a rare, life-threatening condition characterised by abnormal ventilatory response to hypoxia and hypercapnia due to failure of autonomic respiratory control.218 CCHS is not per se a neurocristopathy due to the involvement of both the central and peripheral autonomic nervous system. CCHS patients often present symptoms resulting from a broader dysfunction of the autonomous nervous system and predisposition to neural crest cell derived tumours (5–10% of CCHS cases, neuroblastoma, ganglioblastoma, ganglioneuroma).209 219–221 Haddad syndrome (MIM 209880) is defined by the association of HSCR and CCHS and is found in about 20% of CCHS patients.138 172 173 In these cases, L-HSCR (including TCA) is the most frequent, and the sex ratio is almost equal at variance to what is observed in isolated HSCR.222 PHOX2B is the disease causing gene with de novo heterozygous mutation in the proband,174 the far most frequent being in frame duplication leading to polyalanine expansion.223 224 Parents of patient with molecularly proven CCHS must be tested for accurate genetic counselling as about 10% carry a somatic mosaic137 and some parents may develop late onset CHS.225 Finally, genotype/phenotype correlations allow the detection of patients with a high risk to develop tumours (and carry a frameshift mutation) and reassurance about tumour predisposition to those carrying a polyalanine expansion.137
Waardenburg syndromes (WS) and related pigmentary anomalies
WS, an autosomal dominant condition, is by far the most frequent condition combining pigmentary anomalies and sensorineural deafness (1/50 000 live births and 2–5% of all congenital deafness), resulting from the absence of melanocytes of the skin and the stria vascularis of the cochlea.226 WS is clinically and genetically heterogeneous (MIM 193500, MIM 148820, MIM 193510).227 The combination of HSCR with WS defines the WS4 type (Shah-Waardenburg syndrome, MIM 277580), a genetically heterogeneous condition. Indeed, homozygous mutations of the endothelin pathway118 120 121 168 and heterozygous SOX10 mutations have been identified in WS4 patients.122 Patients carrying a SOX10 mutation may also present with CNS involvement including seizures, ataxia, and demyelinating peripheral and central neuropathies123 228 and WS2 (Bondurand et al in Am J Hum Genet website).
Related syndromes associating pigmentary anomalies and HSCR include: (1) Yemenite deaf-blind hypopigmentation syndrome (MIM 601706), a SOX10 mutation having been reported in one of these families153; (2) Black Locks-Albinism-Deafness syndrome (BADS, MIM 227010) with TCA-HSCR in one case169; (3) aganglionic megacolon associated with familial piebaldism (MIM 172800)170 171; (4) HSCR and profound congenital deafness but with no other WS features has also been reported.229
Other neurocristopathies
Familial dysautonomia syndrome (FDS, Riley-Day syndrome, MIM 223900) has been reported once in association with HSCR. Although it could have arisen by chance alone, it is interesting to note that FDS maps to 9q31 where a susceptibility locus for HSCR has been identified. Other occasional associations reported thus far include cleft lip with or without cleft palate,230 neural tube defects (myelomeningocoele)231 and neurofibromatosis type I.210 The significance of these associations is not yet established.
Other syndromes with HSCR as a frequent feature
Mowat-Wilson syndrome (MIM 235730)
Mowat-Wilson syndrome (MWS) is an MCA-MR condition first delineated among the heterogeneous group of patients with HSCR and MR.134 The condition is associated with microcephaly, epilepsy, a facial gestalt and severe mental retardation. The spectrum of possibly associated malformations is wide and encompasses, in decreasing frequency order, hypospadias, renal anomalies, congenital cardiac defect, agenesis/hypoplasia of the corpus callosum and HSCR.148 193 Heterozygous de novo deletions encompassing the ZFHX1B (zinc finger homeo box 1B) gene or truncating mutations within the gene are found in over 100 cases.135 191 192 Some rare splicing and missense mutations have also been reported.148 Loss of function by haploinsufficiency is the disease causing mechanism in humans. ZFHX1B acts as a transcriptional repressor of smad protein targets and has key functions in early embryonic development in several animal models.94 136 232 A knock-out restricted to NCC precursors in mice demonstrated a wide range of anomalies in NCC derivatives.233
Goldberg-Shprintzen syndrome (MIM 609460).
This autosomal recessive MCA-MR syndrome combines HSCR, moderate mental retardation, microcephaly, polymicrogyria, facial dysmorphic features (hypertelorism, prominent nose, synophrys, sparse hair), cleft palate and iris coloboma.182 183 The disease causing gene KIAA1279 has been identified in a large consanguineous family and encodes a protein of unknown function.184 GSS is a rare condition within the group of patients with MR and HSCR. Several reports with variable association of microcephaly, iris coloboma, cleft palate and mental retardation, and regarded as possible variants GSS, are unlikely allelic conditions.234 235
HSCR with limb anomalies
Several rare syndromes with HSCR and distal limb anomalies (polydactyly or hypoplasia) have been reported. These are: (1) HSCR with polydactyly, unilateral renal agenesis, hypertelorism and congenital deafness (MIM 235740)185; (2) HSCR, postaxial polydactyly and ventricular septal defects (MIM 235750)186; (3) HSCR, hypoplasia of the distal phalanges and nails and mild dysmorphic features (MIM 235760)187; (4) HSCR with preaxial polydactyly, heart defect and laryngeal anomalies (MIM 604211)188; (5) HSCR with brachydactyly type D (MIM 306980)189; (6) HSCR with brachydactyly, macrocephaly and vertebrae anomalies190; (7) BRESHEK syndrome236; and (8) Werner mesomelic dysplasia.205 206
Bardet-Biedl syndrome (MIM 209900) and McKusick-Kauffman syndrome (MIM 236700)
Bardet-Biedl syndrome (BBS) is characterised by progressive pigmentary retinopathy, obesity, hypogenitalism, renal involvement (including cysts, renal cortical loss or reduced ability to concentrate urine), mild mental retardation and postaxial polydactyly of the hands and feet. BBS is genetically heterogeneous with at least 12 loci and 10 genes identified, all involved in ciliary function.237 HSCR has been reported in several BBS cases.194 195 McKusick-Kauffman syndrome (MKKS) is a rare condition, allelic to BBS and characterised by hydrometrocolpos, postaxial polydactyly and congenital heart defect. HSCR is found in 10% of cases.196 Mutations in the MKKS/BBS6 gene, encoding a chaperonin protein, were identified in some BBS patients confirming that both conditions are allelic.238 239 Jeune syndrome, also ascribed to a gene involved in the ciliary function, has been occasionally associated to HSCR.207
Smith-Lemli-Opitz syndrome (MIM 270400)
Smith-Lemli-Opitz syndrome (SLO) is characterised by pre- and postnatal growth retardation and microcephaly, severe mental retardation, facial dysmorphic features, hypospadias and syndactyly between toes 2 and 3. SLO results from cholesterol metabolic impairment with mutation of the 7-dehydro-cholesterol reductase gene (DHCR7, chromosome 11q12-q13).240 241 HSCR is observed in a significant number of severe SLO patients.197
Cartilage-hair hypoplasia syndrome (MIM 250250)
The skeletal dysplasia cartilage-hair hypoplasia syndrome (CHH), first described in the Old Order Amish community, combines metaphyseal dysplasia with short limb dwarfism, fine, sparse and blond hair, transient macrocytic anaemia and immunodeficiency. HSCR is associated in approximately 10% of the cases.198 The gene RMRP has been mapped to chromosome 9p13.242 Interestingly, HSCR has been reported in the Holmgren-Connor syndrome (MIM 211120) which may be allelic to CHH.
The RET gene plays a pivotal role in both isolated and syndromic HSCR. Indeed, epistatic interactions with the common RET hypomorphic allele has been demonstrated for HSCR predisposition in Down, CCHS and BBS.148 243 Conversely, the role of the RET hypomorphic allele is not significant in MWS and WS4 due to SOX10 mutation.148 Of note, a case–control study in a Chinese population identified an SNP in intron 2 of PHOX2B (IVS2+100) as over represented in the HSCR group of patients.244
Miscellaneous observations
This can be include; (1) syndromes with myopathy200 201; (2) syndromes with dermatological findings202; and (3) syndromes with central nervous system anomalies, among which the HSAS/MASA spectrum ascribed to mutations in the X-linked L1CAM gene. Indeed, at least five different mutations in L1CAM have been identified in patients with hydrocephalus and HSCR.199 Interestingly, L1cam is an ENS-expressed gene.29 The question of L1CAM being a modifier gene in HSCR has been raised with no definitive answer given thus far.245 246 Other rare associations include the finding of HSCR with Fryns, Aarskog, Jeune asphyxing thoracic dystrophia, Joubert, frontonasal dysplasia, osteopetrosis, Goldenhar, Lesch-Nyhan, Rubinstein-Taybi, Toriello-Carey, Pallister-Hall, spondylo-epimetaphyseal dysplasia with joint laxity (SEMDJL, MIM 271640), persistent mullerian duct syndromes, and asplenia with cardiovascular anomaly.
Associated anomalies
A wide spectrum of additional isolated anomalies have been described among HSCR cases, with an incidence varying from 5–30% according to series.10 11 13 247–250 No constant pattern is observed and these anomalies include distal limb, sensorineural, skin, central nervous system, genital, kidney and cardiac malformations. However, cardiac defects, and mostly atrio- or ventriculoseptal defects, are found with an incidence of 5% of HSCR cases, once removed patients with trisomy 21 and HSCR. Renal dysplasia or agenesis was reported in FMTC251 and found in 4.4% in a series of 160 HSCR cases and may still be underestimated (personal data). This is of interest since homozygous knock-out mice for genes involved in the Ret signalling pathway present with renal agenesia/dysplasia in addition to megacolon.82 Genital anomalies including hypospadias are reported in up to 2–3% of HSCR patients. Gastrointestinal malformations such as Meckel diverticulum, pyloric stenosis, single umbilical arteria, inguinal hernia or small bowel atresia are also found.252–254 Finally, facial dysmorphic features seem extremely frequent when looked for. These data highlight the importance of a careful assessment by a clinician trained in dysmorphology for all newborns diagnosed with HSCR. Skeletal x ray, cardiac and urogenital ultrasound survey should be systematically performed. The observation of one additional anomaly to HSCR should prompt chromosomal studies and/or molecular karyotyping.
The unanswered question of sex-dependent penetrance in HSCR
Expression and penetrance of a RET mutation is variable and sex-dependent within HSCR families. In large series, the estimated penetrance is 72% in males and 51% in females.44 Accordingly, the penetrance of the HSCR predisposing T allele for the HSCR trait is greater in females than in males.75 A significant parent-of-origin effect at the RET locus, 78% of shared RET alleles by affected sibs being maternally derived, could explain the sex difference in HSCR expression. Epigenetic effects at the RET locus have been hypothesised. Of note, a sex difference in disease expressivity (that is, length of the aganglionic segment) of Ret+/−; Ednrbs/s mice has been observed.255 None of the genome wide scans performed in HSCR families identified a locus on the X chromosome thus far. However, a differential screen for ENS expressed genes performed in mice pointed to several X-linked genes.29 Finally, the observation of a skewed sex ratio in RET-independent syndromic HSCR (that is, MWS and WS4) suggests that the sex bias observed cannot entirely rely on a gender effect at the RET locus.148
GENETIC COUNSELLING
HSCR is a sex-modified multifactorial congenital malformation with an overall recurrence risk in sibs of the proband of 4% (relative risk = 200). In isolated HSCR, adequate relative risk figures will be provided by taking into account the sex and length of the aganglionic segment in the proband and the gender of the sibling (2–33%). According to Carter’s paradox, the highest recurrence risk is for a male sibling of a female proband with L-HSCR (table 1). According to poor genotype–phenotype correlation thus far, the benefit of mutation screening for HSCR patients appears low except for systematic testing of exon 10 and 11 of the RET gene, owing to cancer predisposition of MEN2A mutations. This, however, is still not routine practice in most countries.
Many HSCR cases are associated with other congenital anomalies. In these cases, the long term prognosis is highly dependent on the severity of the associated anomalies. Several known syndromes have straight Mendelian inheritance. This emphasises the importance of careful assessment by a clinician trained in syndromology of all newborns diagnosed with HSCR.
Acknowledgments
We thank the HSCR patients and their families, and the French Hirschsprung Disease Association (AFMAM), for their cooperation and active participation over 10 years. We sincerely acknowledge colleagues from all over the world for providing us samples as well as all the students and collaborators of the groups who joined in a consortium on Hirschsprung disease.
REFERENCES
Footnotes
Competing interests: None declared.