Article Text

Abstract
Many cellular functions depend on the correct delivery of proteins to specific intracellular destinations. Mutations that alter protein structure and disrupt trafficking of the protein (the “cargo”) occur in many genetic disorders. In addition, an increasing number of disorders have been linked to mutations in the genes encoding components of the vesicular transport machinery responsible for normal protein trafficking. We review the clinical phenotypes and molecular pathology of such inherited “protein-trafficking disorders”, which provide seminal insights into the molecular mechanisms of protein trafficking. Further characterisation of this expanding group of disorders will provide a basis for developing new diagnostic techniques and treatment strategies and offer insights into the molecular pathology of common multifactorial diseases that have been linked to disordered trafficking mechanisms.
- AD, Alzheimer disease
- AP, adaptor protein
- APP, amyloid precursor protein
- ARC, arthrogryposis, renal dysfunction and cholestasis syndrome
- BLOC, biogenesis of lysosome-related organelle complex
- CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma
- CMT, Charcot–Marie–Tooth
- COP, coat protein
- ER, endoplasmic reticulum
- ERGIC, ER–Golgi intermediate compartment
- GS, Griscelli syndrome
- GTPase, guanine triphosphatase
- HOPS, homotypic fusion and vacuolar protein sorting
- HPS, Hermansky–Pudlak syndrome
- HS, haemophagocytic syndrome
- HSP, hereditary spastic paraplegia
- IDT, inherited disorders of trafficking
- LAMP, lysosome-associated membrane protein
- LRO, lysosome-related organelle
- OMIM, Online Mendelian Inheritance in Man
- SCA5, spinocerebellar ataxia type 5
- SNARE, soluble N-ethylmaleimide-sensitive fusion attachment protein receptor
- TGN, trans-Golgi network
- VAMP, vesicle-associated membrane protein
- inherited trafficking disorders
- vesicular trafficking
- intracellular protein trafficking
Statistics from Altmetric.com
- AD, Alzheimer disease
- AP, adaptor protein
- APP, amyloid precursor protein
- ARC, arthrogryposis, renal dysfunction and cholestasis syndrome
- BLOC, biogenesis of lysosome-related organelle complex
- CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma
- CMT, Charcot–Marie–Tooth
- COP, coat protein
- ER, endoplasmic reticulum
- ERGIC, ER–Golgi intermediate compartment
- GS, Griscelli syndrome
- GTPase, guanine triphosphatase
- HOPS, homotypic fusion and vacuolar protein sorting
- HPS, Hermansky–Pudlak syndrome
- HS, haemophagocytic syndrome
- HSP, hereditary spastic paraplegia
- IDT, inherited disorders of trafficking
- LAMP, lysosome-associated membrane protein
- LRO, lysosome-related organelle
- OMIM, Online Mendelian Inheritance in Man
- SCA5, spinocerebellar ataxia type 5
- SNARE, soluble N-ethylmaleimide-sensitive fusion attachment protein receptor
- TGN, trans-Golgi network
- VAMP, vesicle-associated membrane protein
It has been known for decades that genetic mutations may cause misfolding and aggregation of the mutant protein. Such aggregates are found most commonly in the endoplasmic reticulum (ER) (Aridor and Hannan1,2 and references within). Abnormal protein-modification processes may also cause generalised intracellular protein mislocalisation in inherited diseases such as congenital disorders of glycosylation and mucolipidosis.
The subject of this review is the increasing number of inherited human diseases in which protein mislocalisation results from mutations in the genes directly involved in vesicular trafficking. Vesicular transport is a process by which membrane-bound vesicles (“carriers”) are released from the donor compartment and travel to a specific cellular location (the acceptor site). On reaching the acceptor, the carrier membrane fuses with that of the target organelle and delivers its contents to their destination. This concept was first introduced by Palade3 and developed by many others.4–6
The process of intracellular protein transport is divided into two major routes depending on its direction in relation to the cell membrane: the biosynthetic and the endocytic pathways.
The biosynthetic or secretory pathway describes the journey of all proteins from the time of their synthesis as polypeptides in the cytosolic ribosomes. These polypeptides undergo folding and sometimes oligomerisation into multisubunit protein complexes in the ER. Once the proteins are ready to leave the ER, they are identified by the cargo-sorting machinery and packaged into the transport vesicles for transport to the Golgi apparatus. En route to the Golgi, proteins congregate at the ER–Golgi intermediate compartment (ERGIC), from where they are delivered to the cis side of the Golgi apparatus. They then move through the Golgi cisternae to the trans-Golgi network (TGN). Post-translational modifications such as glycosylation tend to occur in the cisternae, whereas in the TGN, the proteins undergo final sorting into various routes leading to their destination. For many proteins, post-translational modification will determine their final position in the cell. Although the precise sorting signals are not known in many cases, addition of mannose-6-phosphate to N-glycans allows receptor-mediated sorting of the lysosomal hydrolases to endosomes and then to lysosomes.7 In polarised cells (such as renal tubular epithelial cells) specialised trafficking processes ensure that basolateral and apical membranes maintain their unique protein composition.8,9 Cytoskeleton proteins and associated molecular motors such as kinesin, dynein and dynactin have an important role in transporting vesicles between intracellular compartments (as in a complicated underground communication network, an intracellular mesh of microtubules and filaments allows rapid shuttling of cargo carriers to their correct destination).
In the endocytic pathway, molecules from the extracellular space and plasma membrane are internalised and carried in peripheral early sorting endosomes back to the plasma membrane (eg for recycling of integral membrane proteins) or transferred to late endosomes and then delivered to lysosomes for digestion.10–12 Transcytosis, a trafficking process that is found only in polarised cells, delivers, via vesicular carriers, proteins originating at one plasma-membrane domain to the opposite cell surface. Examples of transcytosis include trafficking of polymeric immunoglobulin receptor, which is delivered from the basolateral to apical membranes in human intestinal epithelial cells and may have a role in the mechanism of food allergy.13
In polarised cells, the regulation of both secretory and endocytic pathways has to be particularly tightly controlled. Hence, although some proteins, such as the clathrin adaptor protein (AP)-2 complex, function in both non-polarised and apical endocytosis, others, such as guanine triphosphatase (GTPase) dynamin, exist in two forms. Factors that influence the choice of the trafficking pathway include the cell type, cargo protein type, modification status and ligand.14 As part of the traffic-regulating mechanism, a large number of GTPases act at all stages of the trafficking progress in the recruitment of effector proteins and ensuring smooth running of the vesicular transport.
Inherited disorders of the vesicular trafficking machinery
The number of known inherited disorders of trafficking (IDT) has increased substantially since this topic was reviewed by Olkkonen and Ikonen.15 Although IDT phenotypes are very variable, frequent features include hypopigmentation, disturbed cell-mediated immune response and neurological abnormalities (table 1). The importance of apical and basolateral trafficking is underlined by the disorders affecting polarised cells such as renal tubular cells, hepatocytes, and cells of the inner ear.
Examples of human disorders caused by the defects in vesicular trafficking machinery
To illustrate how discoveries of the disease-causing mutations have advanced the knowledge of the normal and pathological cellular protein trafficking processes, IDT are considered according to the predicted function of the gene product in the vesicular trafficking pathway. Thus, we have subclassified IDT into: (1) disorders of cargo recruitment and vesicle biogenesis, (2) defects in Rab and other GTPases and associated proteins, (3) abnormalities in the cytoskeleton and cytoskeleton-associated proteins, and (4) defects in vesicular tethering and fusion. In the interests of brevity, only a few illustrative examples from each group are discussed.
Disorders of cargo recruitment and vesicle biogenesis
Various transport membrane-bound vesicles and organelles differ in shape, size and composition of their membrane and luminal contents. Despite this constitutional diversity, vesicles follow similar general rules of intracellular trafficking. Proteins destined to be ferried between the two membrane-bound compartments are actively recruited with the help of ligands and receptors to the location on the membrane where the transporting vesicle is assembled (fig 1A).

Vesicular transport. (A) Trafficking vesicle assembly. Cargo protein displaying sorting signal forms a complex with its specific soluble ligand in the donor compartment. The ligand is recognised by the transmembrane receptor. Vesicular soluble N-ethylmaleimide-sensitive fusion attachment protein receptor protein (v-SNARE) is present in the membrane of the donor compartment. Budding of the vesicle occurs with the aid of coat-protein complexes and Rab proteins, which became active, acquiring GTP with the aid of Rab activator molecules (RA). (B) Intracellular traffic. Vesicles move along the cytoskeletal tracks, either actin filaments or microtubules, with the help of motor proteins, activated Rabs and Rab effector molecules. After Rab inactivation (RI) and dissociation of the protein coat, the vesicle becomes tethered to the acceptor compartment membrane and target SNARE (t-SNARE) proteins. Tethering is achieved by a combination of tethering factors such as RabB and Sec1/Munc18 (SM) proteins. (C) Fusion of the vesicle to the target membrane. During fusion v-SNARE and t-SNARE proteins oppose and change conformation, forming a four-helix bundle. The cargo protein and its ligand are released into the acceptor compartment for further sorting (adapted from Olkkonen and Ikonen15).
The importance of this process in human physiology was illustrated by the discovery of the underlying molecular defect for a combined deficiency of clotting factors V and VIII, an autosomal recessive bleeding disorder characterised by reduced circulating levels of both proteins (OMIM 227300). Patients with this condition have mutations in either ERGIC-53, which is a major constituent of the ERGIC (also known as mannose-binding lectin1 (LMAN1) or multiple coagulation factor deficiency 2 (MCFD2) genes. The cytosolic protein MCFD2 specifically recruits clotting factors V and VIII, and forms a complex with the membrane-associated ERGIC-53 to transport the clotting factors from ER to Golgi in coat protein (COP)II-coated vesicles.16
The process of vesicle biogenesis is mediated by cytosolic multisubunit COP complexes, their adaptors, and other associated proteins that directly interact with transmembrane proteins or receptors for luminal molecules.17–19 COP complexes have the capacity to cause distortion of the membrane shape, which leads to budding and release of coated transport vesicles. Three types of vesicular coats (COPI, COPII and clathrin-containing) have been extensively investigated, although other types of coats undoubtedly exist. COPII proteins have been localised to secretary vesicles involved in traffic from ER to ERGIC, whereas COPI are involved in traffic from ERGIC to Golgi and also function at some steps in the endocytic pathway. Clathrin-coated vesicles can be found predominantly in the endocytic pathway, delivering vesicles from the plasma membrane, endosomes and TGN. Cargo specificity is mediated by different vesicular coats and also coat-associated proteins that recognise cargo-sorting signals, including specific amino acid motifs, saturated fatty acyl moieties and carbohydrates, which are recognised by the lectin-like receptors, and also dynamic lipid domains formed by cholesterol and glycosphingolipids. In polarised cells, proteins in the secretory and endocytic pathways are targeted to their correct domains after segregation of apical and basolateral proteins into distinct carriers. Luminal carbohydrate moieties or lipid-binding properties of transmembrane anchors are often important for inclusion into apical carriers. In contrast, sorting of membrane proteins into basolateral carriers depends on recognition of discrete cytoplasmic domain targeting signals, involving tyrosine or dileucine motifs, many of which are recognised by clathrin adaptor complexes.20 Additional specificity in vesicular formation is provided by inositol phospholipids (phosphoinositides) and small GTPases. It appears that the number of phosphate modifications on the inositol ring determines the site of its action in the vesicular trafficking pathways.21 For example, phosphoinositol 4,5-biphosphate binds AP-2 on the plasma membrane, whereas phosphoinositol 4-monophosphate activates AP-2 at the TGN.
Membrane budding and vesicle release requires lipid-binding scaffold proteins and lipid-modifying enzymes. Thus lipid-binding proteins such as brain-specific AP180 may bind to phosphoinositides and support membrane deformation acting together with clathrin adaptor proteins.22 Amphiphysin, which contains the membrane curvature-sensing BAR domain detects the initial budding of the vesicle and then induces further curvature, ultimately leading to vesicle scission.23
The final step in the vesicle formation (scission from the donor membrane) is mediated by the concerted action of cytoskeleton proteins, lipid-binding proteins and GTPases such as the large GTPase dynamin that directly interacts with actin regulatory proteins.
The COPII coat structure has been delineated in yeast, in which a small GTPase SAR1 interacts with an integral membrane glycoprotein SEC12 on the cytosolic surface of the ER and then binds the SEC23–SEC24 complex responsible for cargo recognition and binding. The final layer in this sandwich is provided by the SEC13–SEC31 complex, which coats the cargo proteins, leading to vesicle formation and budding.
Loss of sec23 function in yeast and worms leads to distension of the terminal portions of the ER, resulting from blockage of the secretory pathway at the ER exit sites.24 A F382L missense substitution in SEC23A, one of two human homologues of the yeast sec23 gene, causes cranio-lenticulo-sutural dysplasia (CLSD; OMIM607812). This autosomal recessive disorder is characterised by delayed ossification of the cranial bones, hyperpigmentation and cataracts. Analysis of patients with CLSD found distended ER cisternae with collagen accumulation in fibroblasts from affected patients, and similar, but less dramatic, changes in fibtoblasts from heterozygotes. It has been suggested that abnormal bone and connective-tissue formation may result from a defect in the secretion of collagen and/or other related extracellular matrix proteins.24 In zebrafish, a truncating L402X mutation in sec23a homologue was discovered to cause the Crusher mutant, which has short body length, small and malformed head skeleton, and absence of cartilaginous ear capsules.25 Electron microscopy revealed that Crusher chondrocytes accumulated extracellular matrix proteins (particularly collagen2a1) in the distended vacuolar compartments of the ER and also displayed abnormally formed Golgi apparatus. There was absence of glycosylation of the matrix proteins, underlining the defect of Golgi transport. Morpholino knockdown of the second SEC23 homologue, sec23b, in zebrafish caused complete loss of the ventral pharyngeal skeleton and abnormalities of the neurocranium. Thus, it appears that neurocranium is particularly sensitive to the abnormalities in the pathway of COPII vesicle biogenesis, and that both SEC23A and SEC23B are required for normal chondrocyte activity and craniofacial skeletal development.
Mutations in the genes encoding the subunits of the AP-3 complex cause Hermansky–Pudlak syndrome (HPS) type 2 (OMIM 608233), and the characteristics of the pearl and mocha mouse strains. HPS comprises a group of autosomal recessive disorders characterised by oculocutaneous albinism and bleeding tendency, and is caused by mutations in at least eight human genes. Mutations in 16 different mouse genes result in HPS-like mouse phenotype (Gautam26 and references within). HPS is characterised by a defect in the biogenesis of multiple lysosome-related organelles (LROs), including melanosomes, platelet secretory granules, lamellar bodies of type II alveolar epithelial cells, and lytic granules of cytotoxic T lymphocytes and natural killer cells.27 LROs originate from small vesicles transporting protein cargo through a complex endosomal system. They gradually develop into mature organelles that share some of their biophysical characteristics, such as acidic pH, with lysosomes. In a budding vesicle, the AP-3 complex forms part of a heterogeneous membrane-proximal layer onto which clathrin coat is deposited. It is also important for recognition of the sorting signals for transport of integral membrane proteins from endosomes to lysosomes and LROs.28 The cargo-specific function of AP-3 is shown by melanocytes: AP-3 is required for normal trafficking of tyrosinase, but not of tyrosinase-related protein 1, to maturing melanosomes.29
Most other HPS-related proteins belong to three distinct protein complexes, the biogenesis of lysosome-related organelle complex (BLOC) proteins I–III. Although the exact molecular function of these complexes is unknown, their binding partners include soluble N-ethylmaleimide-sensitive fusion attachment protein receptor (SNARE) proteins such as syntaxin 13, dystrobrevins, tubulin and actin filaments, implicating BLOC proteins in LRO biogenesis and trafficking (Dell’Angelica30 and references within).
Defects in Rabs and Rab-associated proteins
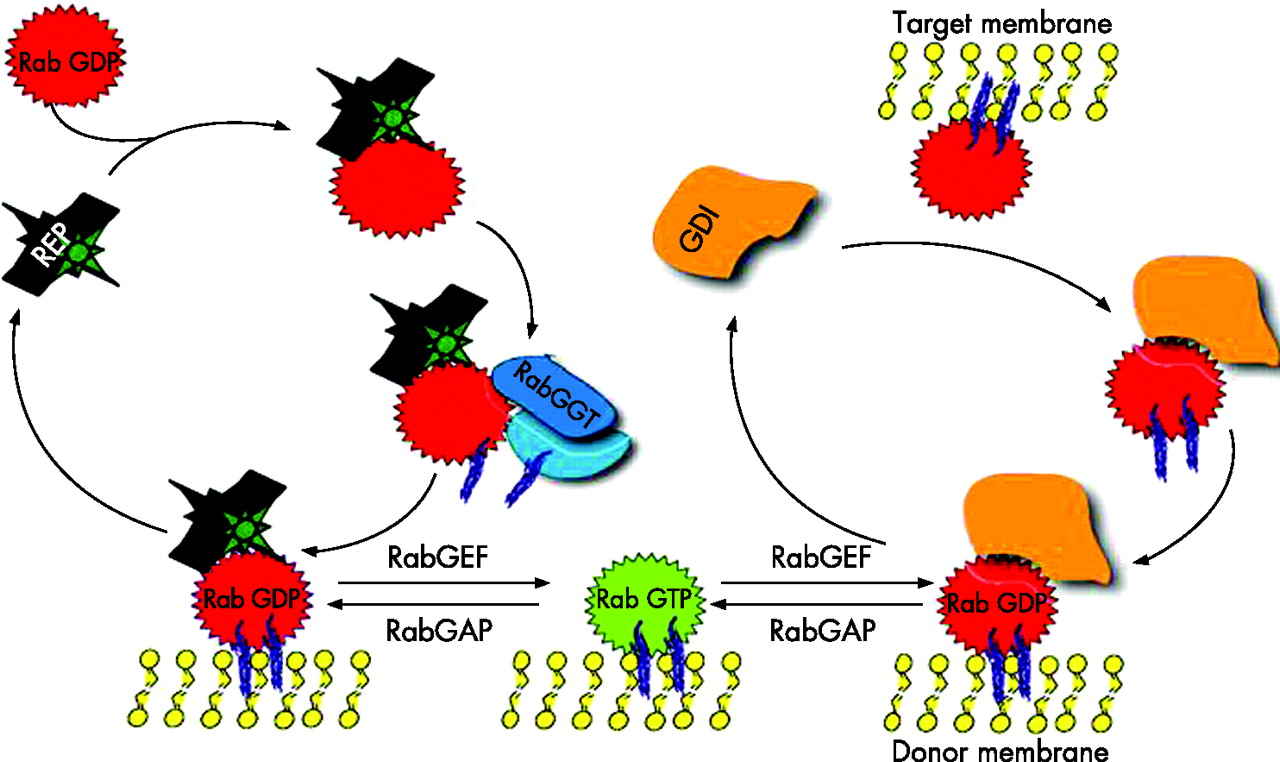
Rab proteins are small GTPases of the Ras superfamily and are present in all eukaryotes. At least 60 human RAB genes are known, and their function is essential for the regulation of multiple steps of intracellular membrane trafficking (vesicle budding, movement, tethering and fusion) (fig 1B). Like all GTPases, they change their conformation between the active (GTP-bound) and inactive (GDP-bound) form. The balance between the two isoforms is finely regulated by Rab GTPase-activating proteins and Rab guanine nucleotide exchange factors. When activated by GTP binding, they recruit step-specific effector proteins, which may have a role in linking vesicle and target membranes to the cytoskeleton.31 Effectors dissociate when Rabs convert to the GDP-bound state, and the inactive Rab can be removed from the membrane by the Rab GDP dissociation inhibitor (fig 2).

Rab cycle (see text for details). REP, Rab escort protein; RabGGT, Rab geranylgeranyl transferase; GDI, Rab GDP dissociation inhibitor; RabGEF, Rab guanine nucleotide exchange factor; RabGAP, Rab GTPase activating protein (adapted from Seabra33).
Post-translational isoprenylation (geranylgeranylation) of Rab GTPases is essential for their membrane association. This reaction is catalysed by Rab geranylgeranyltransferase and is assisted by the Rab escort proteins, which form stable complexes with newly synthesised GDP-bound Rabs and deliver them to the target membrane (fig 2).32
Abnormalities in several Rabs and Rab-associated proteins are known to cause human diseases and mutant mouse models.33 Mutations in the gene encoding Rab27A causes type II Griscelli syndrome (GS; OMIM 607624), an autosomal recessive condition characterised by hypopigmentation of the skin, a silvery-grey sheen to the hair, the presence of large clumps of pigment in the hair shaft, and an abnormal accumulation of end-stage melanosomes in the centre of melanocytes.34 Three clinical types of GS (I–III) have been described. All three have typical dermatological characteristics, but there are important clinical distinctions between the types. Patients with RAB27A mutations (GS-II) develop haemophagocytic syndrome (HS), characterised by episodes of life-threatening uncontrolled T lymphocyte and macrophage activation. During episodes of HS, activated T cells and macrophages infiltrate various organs (including the brain), leading to massive tissue damage, organ failure and death in the absence of immunosuppressive treatment. Bone-marrow transplantation is curative for this condition.35 Expression of Rab27A is high in melanocytes, cells of the haematopoietic system and other secretory cells, and is involved in the control of lytic granule release.
Patients with GS-I (OMIM 214450) have severe primary neurological impairment in association with a typical pigmentary abnormality. Patients exhibit severe developmental delay and mental retardation but have no immune dysfunction.36 This variant of GS is caused by mutations in the MYO5A gene encoding the actin-binding organelle motor protein myosin VA, which supports movement of melanosomes and lysosomes.37,38
Identical pigmentary changes in GS1 and GS2 suggest that myosin VA and Rab27a should have complementary functions but they do not appear to interact directly. However, a direct interaction was found between the SHD domain of SLAC2/melanophilin (Mlph) and Rab27a, and between the C-terminal domain of Mlph and the globular tail of myosin Va.39 Thus Rab27a/Mlph/MyoVa complex is essential for the capture and local transport of melanosomes.40,41
In addition, mutations in the MLPH gene encoding Mlph have been found in patients with GS-3 (OMIM 609227).42 Patients from this subgroup have the pigmentary characteristics of GS but no additional features.
GS demonstrates how different interacting partners provide diversity to the function of the same component of the trafficking machinery. Although the presence of all three (Rab27a, Myo5a and Melanophilin) proteins is crucial for the trafficking of melanosomes, neurodevelopmental abnormalities are specific for Myo5a deficiency, and Rab27a uses a different effector for its function in cytotoxic T lymphocytes. Expression of Rab27a is high in melanocytes, haematopoietic tissues, retinal pigment epithelium and pancreatic islet cells, but not in brain. In retinal epithelium, Rab27a forms a complex with the MyRIP and Myo7 proteins, and by linking them to the actin cytoskeleton, assists with melanosome transport. In pancreatic β-cells, it mediates the transport of the secretory insulin-containing granules and may be involved in increasing insulin secretion.43,44 A defect of lytic granule exocytosis is found in the ashen (Rab27a-negative) mouse and in patients with GS-II. Rab27a was found to interact directly with Munc13-4 protein, a positive regulator of degranulation of lytic granules.45 Munc13-4 is a peripheral membrane protein localised to the lytic granules, and recently mutations in the gene encoding Munc13-4 were found to cause one of the types of HS (OMIM 608898).46
The importance of Rab proteins in brain development and function is emphasised by the discoveries of the involvement of Rab3-associated proteins in human disease, and that mutations in RAB23 and RAB 3A result in the opb and earlybird mutant mouse strains, respectively. The earlybird mouse displays behavioural abnormalities including disturbance of circadian rhythms and sleep homeostasis, whereas opb mice die during the second half of gestation, and have an open anterior neural tube in the head and spinal cord, abnormal somites, polydactyly and poorly developed eyes.47 Rab23 localises to early lysosomes and its action is linked to the sonic hedgehog (shh) pathway. Rab23 is expressed at high levels in mouse spinal cord (especially the dorsal half), somites, limb buds and cranial mesenchyme at E10.5 days. Mutations in shh and opb cause opposing transformations in neural cell fate: shh mutant embryos lack ventral cell types throughout the spinal cord, whereas opb mutant embryos lack dorsal cell types, specifically in the caudal spinal cord. It appears that opb may act downstream of shh. Ventral cell types that are absent in shh mutants are present in shh-opb double mutants. Thus dorsalising signals may activate transcription of Rab23 to silence the shh pathway in dorsal neural cells.47,48
Rab3A is the most abundantly expressed Rab protein in the brain, where it is present in virtually all synapses and is involved in Ca2+-dependent neurotransmitter release.49 It also has high expression in pancreatic islet cells and may have a negative role in insulin secretion.43,50 This role appears to be mediated by granuphilin, which also interacts with Rab27a.43 It is interesting to note that in this case two Rab proteins appear to have the opposite effect on insulin secretion mediated by the same effector. Mutations in two genes implicated in Rab3a regulation are associated with neurodevelopmental defects. Mutations in the gene encoding Rab GDIα, which binds Rab3A, cause X-linked mental retardation, whereas recessively inherited changes leading to defects in the catalytic and non-catalytic subunits of Rab3GAP cause Warburg micro syndrome (WMS; OMIM 600118) and Martsolf syndrome (OMIM 212720), respectively.51–53 WMS is a severe autosomal recessive disorder characterised by developmental defects of the eye (micro-ophthalmia and cataract) and brain (microcephaly, cortical gyral abnormalities and hypoplasia of the corpus callosum) and microgenitalia. Martsolf syndrome is characterised by a similar but milder phenotype. Rab3GAP specifically converts active Rab3-GTP to inactive Rab3-GDP and thus may determine the timing of dissociation of Rab3a from synaptic vesicles. The neuronal migration defects and microgenitalia may be secondary to abnormal vesicular secretion of neurotransmitters and of hormones produced by the hypothalamic/pituitary axis. In concordance with this hypothesis, rab3gap gene knockout mice accumulate the GTP-bound form of Rab3A in the brain, which leads to the inhibition of Ca2+-dependent glutamate release from cerebrocortical synaptosomes.54 Those authors suggested that abnormality in exocytosis is due to suppressed dissociation of the Rab3a from the synaptic vesicles, resulting from abnormal conversion of GTP-Rab3a to GDP-Rab3a.54
Cell-biology research identifies new roles in addition to those already known for Rab and Rab-associated proteins in intracellular traffic. For example, Rab11a and MyoVb were found to act as regulators of the apical membrane polarisation in the liver, thus underlying their potential importance in the pathogenesis of human cholestatic disorders.55
Abnormalities in the cytoskeleton and cytoskeleton-associated proteins
A large number of human disorders affect cytoskeleton-associated trafficking. Most of these disorders are associated with neurodegeneration.56,57 The formed cargo-containing vesicle is guided towards the target membrane by the cytoskeleton structures such as microfilaments and microtubules. Microtubules are polymers composed of α and β tubulins, which have a fast-growing peripheral “plus” end and usually a centrally located, slow-growing “minus” end (fig 3). In interphase, microtubules are seen to radiate from the microtubule-organising centre located near the nucleus. Hereditary spastic paraplegias (HSPs) are a group of disorders linked to the abnormalities of the traffic along microtubules and are characterised by progressive lower limb spasticity and weakness.58 Patients with HSP display progressive degeneration of the distal ends of long spinal neurones in the corticospinal tracts. Mutations in the gene SPG4 encoding spastin cause the commonest form of autosomal dominant HSP (OMIM 182601), with a mean age of onset of 20–39 years.59 In addition to the features of retrograde axonopathy caused by corticospinal tract degeneration, patients have cognitive impairment and may also have a thin corpus callosum and cerebellar atrophy.60,61 Spastin is an AAA ATPase, the N-terminal portion of which contains an MIT domain found in microtubule-interacting and trafficking molecules, and binds CHMP1b protein which is associated with the ESCRTIII (endosomal sorting complex required for transport) complex.62,63 SPASTIN was found to be homologous to the microtubule-severing AAA ATPase katanin, and its function in microtubule remodelling appears to be crucial for axonal maintenance.64 Defects in the MIT domain-containing protein SPARTIN occur in Troyer syndrome (OMIM 275900), an autosomal recessive HSP, and microtubule-associated kinesin family motor protein KIF5A mutations occur in both early-onset and late-onset HSPs (OMIM 604187).65–67 Thus microtubule-related transport is critical for upper motor neurone maintenance and function.68

{kind=link}
{kind=link}
{kind=link}
Axonal traffic (see text for details). ES, endosomes; MT, microtubules; N, nucleus; SV, synaptic vesicle (adapted from Hirokawa and Takemura6).
Cytoskeletal motors such as kinesin superfamily proteins (KIFs) and cytoplasmic dyneins are linked to the microtubules by the dynactin complex, and carry various cargos, including membrane-bound organelles such as endosomes or synaptic vesicles, protein complexes and mRNAs, along microtubules.68 KIF family proteins show marked similarities in their motor domain but the rest of the protein structure is extremely diverse, probably to allow for the cargo specificity. A mutation in the KIF1B gene was found in a family with autosomal dominant Charcot–Marie–Tooth (CMT) disease type 2A (OMIM 605995).69 CMT constitutes a clinically and genetically heterogeneous group of diseases affecting peripheral nerves, leading to peripheral muscle weakness and atrophy, sensory loss and depressed tendon reflexes. CMT is classified into two types: demyelinating (type 1) and axonal (type 2) (although the primarily demyelinating defects can develop into functional axonopathies). Kif1a and Kif1b knockout mice show motor and sensory nerve defects, and reduced numbers of synaptic vesicles at the synaptic terminals. KIF1B encodes two isoforms: KIF1Bα, a conventional mitochondrial motor protein, and KIF1Bβ, which has been implicated in transporting synaptic vesicle precursors. In one study, a KIF1Bβ mutation impaired the motor activity of the protein and led to abnormal transport of synaptic vesicle precursors.69 The phenotype of patients with KIF1Bβ mutations overlaps with that of patients with CMT with mutations in RAB7 (OMIM 608882) suggesting that the two proteins may be involved in the same pathway.70,71 In addition to axonal transport, KIF-dependent cellular trafficking has also been demonstrated to be important for ciliogenesis, left–right axis establishment, and maintenance of cell polarisation.72
Elements consisting of actin and spectrin connect transport vesicles to microtubules.73 In particular, spectrin has an important role in linking dynein and dynactin to the vesicular membrane phospholipids. Mutations in BETA-III SPECTRIN cause the slowly progressive form of spinocerebellar ataxia (SCA5, OMIM 604985) characterised by cerebellar cortex atrophy and Purkinje cell loss.74 β-III SPECTRIN localises to the Golgi and vesicular membranes, binds the dynactin subunit ARP1 and stabilises the synaptic membrane localisation of the Purkinje cell-specific EEAT4 glutamate transporter.75,76 Thus in SCA5, β-III SPECTRIN deficiency leads to cargo-specific failure to maintain synaptic membrane protein composition, which may lead to abnormal glutamate signalling in Purkinje cells.74
Unconventional myosins are actin-based cell motors.77 At least nine unconventional myosins are localised in the mammalian inner ear to maintain the highly specialised transport in these polarised epithelial cells, resulting in an extremely well-tuned instrument. Mutations in several unconventional myosins and also γ and β actins lead to sensorineural hearing loss with or without additional abnormalities (Reiners et al78 and references within). In addition, the role of unconventional myosins in auditory sensory hair cells has been investigated in model organisms. Studies of Myosin 6 zebrafish mutants suggested that myosin 6 is required for maintaining the integrity of the apical surface of hair cells, and analysis of the myosin 15 deaf mouse mutant (shaker2) suggested that myosin 15a is essential for the graded elongation of stereocilia in an organised pattern.79,80
Defects in vesicular tethering and fusion
In the final step of its journey, the transport vesicle sheds its coat, moves close to the target membrane and eventually fuses with it. In the past decade, SNAREs have been identified as the proteins central to membrane fusion. Most SNAREs are C-terminally anchored transmembrane proteins that form tight four-helix bundles (SNARE complex) using their N-terminal domains, which pull opposing membranes together.81 The organisation of the nerve-terminal SNARE complex has been extensively investigated. In its formation, two helices are contributed by the SNAP25 SNARE, which is anchored to the target membrane via covalently linked palmitate groups. Two other helices are provided by a vesicle SNARE (vesicle-associated membrane protein (VAMP)), and a target membrane SNARE (syntaxin) (fig 1C). The energy provided by the assembly of the four-helix bundle of the SNARE complex is essential to bring two apposing membranes close enough to fuse.82 Dissociation of the core complex is mediated by recruitment of the NSF ATPase chaperone and α-SNAP, followed by ATP hydrolysis, thereby allowing the components to recycle for another round of membrane fusion. SNARE proteins are ubiquitous and involved in most membrane fusion events, and only a few genetic conditions have been recognised to result from mutations in genes encoding known SNARE proteins.
A homozygous mutation in the SNAP29 gene was described in CEDNIK (cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma, OMIM 609528) syndrome.83 Affected patients present during the first 4 months of life with failure to thrive, hypotonia and abnormal eye movements. Other features include microcephaly, ichthyosis, sensorineural deafness, optic disc hypoplasia, facial dysmorphism and intracranial abnormalities. Skin biopsy shows clear vesicles in several layers, with retained glycosphingolipids, suggesting abnormal lamellar granule maturation.
SNAP29 is homologous to SNAP25 in that it contains two predicted coiled-coil regions that can participate in the formation of the core complex; however, it lacks the palmitoylated membrane-attachment domain found in SNAP25 and localises to intracellular membrane structures (unlike SNAP25, which localises to plasma membrane). It was found that SNAP29 can associate with syntaxin-6 and may have a role in the intracellular trafficking of the IGF1 receptors and other proteins in neuroectodermal tissues.83
Mutations in human SYNTAXIN-11 gene, encoding a t-SNARE STX11 protein, were detected in patients with a form of autosomal recessive familial haemophagocytic lymphohistiocytosis (OMIM 603552), which may also be caused by RAB27A or MUNC13-4 mutations.84,85 STX11 is an integral membrane protein localised to late endosomes and the TGN.86 Interestingly, another form of familial haemophagocytosis, HPLH2 (OMIM 603553), is caused by mutations in the PERFORIN-1 (PRF1) gene, one of the major resident proteins of the cytolytic granules.87
For the SNARE complex to assemble, the upstream regulators or tethering factors have to position the two membranes to oppose each other. This function is performed by protein complexes, which interact with SNAREs, Rab proteins and also Sec1/Munc18 (SM) proteins.88 Although the structure of SNAREs provides some specificity to the membrane-fusion events, tethering factors and other SNARE-associated proteins are crucial for the precision and targeting of the individual vesicles.
ARC (arthrogryposis, renal dysfunction and cholestasis syndrome, OMIM 208085) is an autosomal recessive disorder caused by mutations in the VPS33B gene.89 Additional features of this condition include ichthyosis, platelet α-granule biogenesis defect, severe failure to thrive, recurrent infections and death in infancy. Human VPS33B is an SM protein homologue of the yeast Vps33p, which, together with Vps11, Vps16, Vps18, Vps39 And Vps41 forms the HOPS (homotypic fusion and vacuolar protein sorting) complex. HOPS complex proteins involved in vacuolar biogenesis in yeast and their mammalian homologues are essential for the biogenesis of various LROs such as melanosomes and platelet α-granules.90,91 The HOPS complex has been found to participate in membrane tethering and fusion events at several steps in the endocytic, and possibly secretory, pathway. Two Vps33p homologues, named Vps33a and Vps33b, exist in all multicellular organisms, and overexpression of the A or B homologues appears to induce aggregation of different lysosome-associated membrane protein (LAMP)-1-positive organelles, depending on the tissue type.92 Therefore, it is possible that variations in subunit composition could contribute to the observed functional diversity of mammalian HOPS and to specificity of SNARE-complex formation. Immunostaining of liver and kidney biopsies from patients with ARC identified abnormal localisation of the integral apical membrane proteins polarised cells (hepatocytes and renal tubular cells), suggesting that the defect in polarised trafficking of plasma membrane proteins could be the underlying mechanism for disease development in ARC.89,92
Discovery of mutations in the gene encoding Dysferlin in patients with two types of autosomal recessive limb girdle muscular dystrophy (LGMD2b, OMIM 253601) and Miyoshi myopathy (OMIM 254130) and identification of Dysferlin function in striated muscle cells elucidated a new pathogenic mechanism in the aetiology of muscle disease.93–96 Muscle dystrophies constitute a diverse group of disorders, but most of the genetic defects implicated are related to the function of the dystrophin–glycoprotein complex (DGC), which connects the extracellular matrix to the intracellular actin cytoskeleton and maintains the structural integrity of the skeletal muscle. Skeletal muscle is highly susceptible to damage, owing to the physical stress during contraction, and an efficient plasma-membrane repair mechanism is essential for cell survival. Patients with abnormalities in DGC-related proteins develop significantly more stress-related damage to the sarcolemma. In contrast, dysferlin-null mice do not have any increase in stress-related muscle cell damage compared with controls, but they lose ability to repair damaged plasma membranes.96 In human skeletal muscle, dysferlin is localised to the plasma membrane and cytoplasmic vesicles. It contains a transmembrane domain and six C2 domains shared with other proteins involved in vesicle fusion such as synaptotagmin and Munc13. C2 domains can interact with negatively charged phospholipids and proteins in a calcium-dependent manner. Synaptotagmins promote SNARE-complex formation by binding to SNARE proteins such as syntaxins, SNAP25 and synaptobrevins. It appears that dysferlin-containing vesicles are attracted by an influx of calcium ions to the site of damaged muscle cell membrane, where they fuse to each other and plasma membrane, thus filling the gaps in the sarcolemma.96,97 It was recently found that dysferlin may also have a role in maintenance of the renal podocytes and possibly endothelial cells in the central nervous system.98,99 Interestingly, mutations in another fer-1 homologue encoding the C2 domain protein otoferlin cause human autosomal recessive deafness (OMIM 601071).100 Otoferlin is essential for synaptic vesicle exocytosis and neurotransmitter release in the auditory inner hair cells.101
Links between single gene IDT and common diseases
The identification of the genetic basis for rare inherited vesicular trafficking disorders may also provide insights into the aetiology of some common human diseases. A number of common multifactorial conditions have been linked to abnormalities in vesicular trafficking. Abnormal trafficking of the insulin-sensitive glucose transporter GLUT4 was found in patients with type 2 diabetes. GLUT4 accumulated in the dense membrane compartments, suggesting that defects in membrane trafficking may be involved in the pathogenesis of insulin resistance.102,103 Further delineation of the GLUT4 trafficking pathway may suggest new treatment targets.104
Alzheimer disease (AD) is the commonest cause of dementia in adults. Deposition of senile plaques composed of 38–43 amino acid β-amyloid (Aβ) peptides and hyperphosphorylated tau in neurofibrillary tangles is pathognomonic of the condition.105,106 Aβ is generated by sequential cleavage of amyloid precursor protein (APP) with α, β, and γ-secretases. Familial early-onset dementias are associated with dominantly inherited mutations in genes encoding APP (OMIM 104300, 605714 and 609065) and components of γ-secretase presenilins 1 and 2 (PS-1, OMIM 104311 and PS-2, OMIM 606889),which increase the proportion of Aβ42 among generated Aβ peptides. APP is a transmembrane protein that undergoes glycosylation in the ER and Golgi during its traffic along the secretory pathway. It eventually reaches its location in the dendritic plasma membrane and undergoes recycling in the endocytic pathway. Abnormalities in the vesicular trafficking of APP have been identified in the early stages of the AD and may be critical for the disease pathogenesis.107 The ApoE receptor LR11 may promote trafficking of the APP to multivesicular bodies, which are the likely sites for APP cleavage and generation of Aβ42, and the ApoE receptor pathway may prove to be a target for therapeutic intervention in sporadic early-onset AD (Offe et al108 and references within). Additionally, some authors have suggested that APP acts as a cargo receptor and interacts with the light chain of kinesin-1 in the presence of JIP1b binding, thus mediating anterograde axonal transport for some proteins. However, this hypothesis remains controversial.109,110
Mutations in the microtubule-associated protein tau have been found in several forms of dementia including, familial multiple system tauopathy with presenile dementia (OMIM 600274) and Pick’s disease (OMIM 172700).111 Tau is involved in microtubule assembly and stabilisation, and this function may be compromised by hyperphosphorylation by GSK3β and ubiquitination.112 Tau-positive filamentous inclusions (“neurofibrillary tangles”) in neurons, oligodendrocytes and astrocytes are found in a number of sporadic and familial neurodegenerative disorders.113 Thus, tau dysfunction and secondary abnormalities in trafficking along microtubules can be one of the common mechanisms in neurodegeneration.
CONCLUSIONS
Identification of further genes involved in the intracellular trafficking processes and success in elucidation of the cellular pathogenesis of IDT should lead to opportunities to develop new treatments. Although currently no specific treatment is available for these disorders, chemical chaperones that improve trafficking of misfolded proteins have been used in some conditions in which mutations disrupt the trafficking of the protein product (cargo).114 This approach will inevitably be tested in the diseases with abnormalities in trafficking machinery. The success of these and other approaches to treatment such as development of gene therapy may lead to an improved outlook for these patients. Although IDT are clinically heterogeneous and represented in many organ-based clinical specialities, the identification of shared pathogenetic mechanisms in IDT and the discovery of therapeutic agents active in a variety of IDT might provide a basis for the development of a new cadre of specialists in trafficking medicine, focusing on the diagnosis and management of IDT.
REFERENCES
Footnotes
-
Published Online First 25 May 2007
-
Funding: PG is a GSK Clinician Scientist.
-
Competing interests: None declared.