Article Text

Abstract
Facioscapulohumeral muscular dystrophy (FSHD), an autosomal dominant disorder, represents the third most common human muscular dystrophy. The FSHD disease locus, at chromosome 4q35, is associated with large contractions of the polymorphic repeat sequence array D4Z4. In addition to FSHD disease association with large D4Z4 deletions, a biased interaction with a specific 4qter subtelomeric sequence has been described in patients. Two distinct 4qter subtelomeres, defined as types 4qA and 4qB, have been identified and shown to be equally prevalent in the Caucasian population. In almost all 4q35-linked patients with FSHD, however, disease expression only occurs when large D4Z4 deletions are located on 4qA-defined 4qter subtelomeres. Conversely, large D4Z4 repeat contractions situated on 4qB-defined subtelomeres either are not disease-causing or exhibit a greatly reduced disease penetrance. This study was initiated to confirm this direct FSHD disease association data by measuring the frequency of type 4qA-defined and 4qB-defined subtelomeric sequences in a large cohort of 164 unrelated patients with FSHD from Turkey and the UK, all known to have large D4Z4 deletions. An almost complete association was found between large D4Z4 repeat array deletions located on 4qA-defined 4qter subtelomeres and disease expression in our large FSHD patient cohort. The observed failure of probes 4qA and 4qB to hybridise to two patient-derived DNA samples confirms the presence of an additional rare type of 4qter subtelomeric sequence in humans.
- FSHD, facioscapulohumeral muscular dystrophy
- LGE, linear gel electrophoresis
- PFGE, pulsed-field gel electrophoresis
Statistics from Altmetric.com
- FSHD, facioscapulohumeral muscular dystrophy
- LGE, linear gel electrophoresis
- PFGE, pulsed-field gel electrophoresis
Facioscapulohumeral muscular dystrophy (FSHD), an autosomal dominant muscle disorder in which patients display a progressive weakness of the musculature of the face, shoulders and upper arms,1 is the third most common human muscular dystrophy. FSHD has an estimated incidence of at least 1:20 000 livebirths and exhibits a high level of new mutations, which represent 10–30% of all new FSHD cases.2 A high degree of intra-familial and inter-familial clinical variability is exhibited by patients, with affected individuals often expressing a range of clinical phenotypes, from severe neonatal-onset forms, through “classical” FSHD, to asymptomatic or non-penetrant gene carriers, sometimes all within the same family.3
The FSHD locus is located at 4q35 adjacent to the 4qter subtelomeric region.4 A variable number tandem repeat polymorphic locus, designated D4Z4, was identified in this region, which in normal individuals usually contains multiple integral copies (11>100) of a 3.3 kb KpnI tandem repeat sequence.5 However, in the majority of patients with FSHD, large integral deletions of individual KpnI repeats reduce the size of this D4Z4 repeat arrays down to less than 11 repeats (⩽38 kb). No specific FSHD gene has yet been identified, nor has the underlying pathological mechanism(s) involved in disease expression been explained. An approximate inverse correlation has been reported6,7 between the exact number of D4Z4 repeats retained by a patient and the level of disease severity exhibited by that patient, although the clinical variability associated with FSHD may often confound this relationship.
Two distinct 4qter subtelomeres, designated 4qA and 4qB, have been recognised8,9 and are identified by hybridisation of specific 4qA and 4qB probes to genomic sequences located immediately distal of the D4Z4 repeat array. The 4qA-defined 4qter subtelomere contains an ∼8 kb β-satellite sequence (fig 1) that shows significant sequence homology to the 10qter subtelomere, and hybridisation with the 4qA probe identifies genomic sequences from both subtelomeric regions. The 4qA-defined and 4qB-defined 4qter subtelomeres are found to occur with almost equal frequency in the Caucasian population.8 However, essentially all patients with FSHD exhibit highly biased 4qA/4qB allelic frequencies, in which disease expression is not only associated with large D4Z4 contractions, but that these small (⩽38 kb) D4Z4-containing EcoRI/BlnI fragments must be specifically located on a 4qA-defined 4qter subtelomere.8,9 Conversely, small EcoRI/BlnI fragments that are located either on a 4qB-defined 4qter subtelomere or on a 4qA-defined 10qter subtelomere are either not disease-associated, or they show a greatly reduced disease penetrance.10 In a very small number of DNA samples analysed, both from patients with FSHD and from normal individuals, the small 4q35-derived D4Z4-containing HindIII fragments failed to hybridise to either probe 4qA or 4qB, indicating the possible presence of an additional 4qter subtelomeric type in humans.11

Genomic map of the facioscapulohumeral muscular dystrophy locus region containing 4qA-defined and 4qB-defined 4qter subtelomeres. A schematic map of the 4qter subtelomeric region. The location of the highly variable D4Z4 repeat array, the position of the p13-E11, p4qA and p4qB-defined sequences, the location of the relevant restriction enzyme sites and the position of ∼8 kb β-satellite sequence that defines the 4qA-containing 4qter subtelomere are shown. The large D4Z4-containing HindIII fragment identified by p4qA and p4qB hybridisation is also shown.
This large patient study was therefore undertaken to confirm that it is only the large D4Z4 deletions located on a 4qA-defined 4qter subtelomeres that are associated with FSHD disease expression. The study involved the analysis of DNA samples from 164 unrelated affected individuals, along with a smaller number of normal individuals. Each of the patients with FSHD involved in the study was known to carry a small, disease-associated, 4q35-derived EcoRI/BlnI fragment, as identified by hybridisation to probe p13-E11. Probes 4qA and 4qB were hybridised to HindIII-digested genomic DNA, from patients and controls, separated either by pulsed-field gel electrophoresis (PFGE) or by standard linear gel electrophoresis (LGE), to determine the 4q35-located 4qA/4qB allele frequencies and to correlate this information with the presence of small, 4q35-located D4Z4 repeat arrays.
PATIENTS AND METHODS
FSHD patients
The FSHD patient cohort contained DNA samples from 164 unrelated affected individuals, either obtained for FSHD research purposes or from those patients referred to our molecular diagnostic facility for FSHD analysis. Most patients were from the UK (144), with 15 patients from Turkey and five from Australia. The patients included in the study all conformed to accepted FSHD clinical criteria.
High molecular weight DNA (HMW-DNA) was available for PFGE from 15 Turkish and 50 British patients with FSHD, and from 15 Turkish and 35 British normal individuals. Standard DNA, suitable for linear gel electrophoretic analysis, was available from a further 99 unrelated patients with FSHD, of which 35 were familial disease cases. Control DNA from 50 normal individuals was also available.
All the FSHD patients had previously undergone DNA diagnosis and had been identified as carrying small (∼38 kb, ⩽11 repeats), 4q35-located D4Z4 repeat arrays, as determined by p13E-11 hybridisation to EcoRI-digested and EcoRI/BlnI-digested genomic DNA.12
DNA isolation, analysis of D4Z4 repeat size and 4qA and 4qB variant determination
Standard and high molecular weight DNA was isolated from peripheral blood lymphocytes from patients with FSHD and controls using previously published methods.13 For PFGE analysis, 5 μg HMW-DNA was used for each restriction digest. The 4qA-defined and 4qB-defined subtelomeric alleles were identified by the sequential hybridisation of the probes p4qA and p4qB to HindIII-digested DNA separated on a 0.4–0.5% agarose gel electrophorised at 28 V for 72–48 h and blotted on to hybond membrane.14
RESULTS
All 164 unrelated patients with FSHD were previously identified as having small (⩽38 kb), 4q35-located EcoRI/BlnI fragments. No evidence for somatic mosaicism involving this small EcoRI/BlnI fragment was found in any of the unaffected parents of affected sporadic patients with FSHD who were fully investigated. In many of these de novo cases, only the patient’s DNA was available for a complete screen with p13-E11, and the 4qA and 4qB probes. Xap1 digests of the DNA samples were not routinely carried out, precluding determination of 10qA allele frequencies.
DNA samples from normal individuals, of both Turkish and UK origin, were all found to display equivalent frequencies for the 4q35-located 4qA and 4qB markers, with 53% 4qA and 47% 4qB alleles found overall. The study also showed that either HindIII-digested HMW-DNA analysed by PFGE or HindIII-digested standard DNA analysed by LGE was equally proficient in resolving 4qA-specific and 4qB-specific bands. The increased gel-band resolution that is achievable by PFGE-based analysis permits much greater accuracy when sizing the different DNA restriction fragments on the gel, hence this system is the method of choice routinely used for FSHD DNA analysis in the molecular diagnostic laboratory.
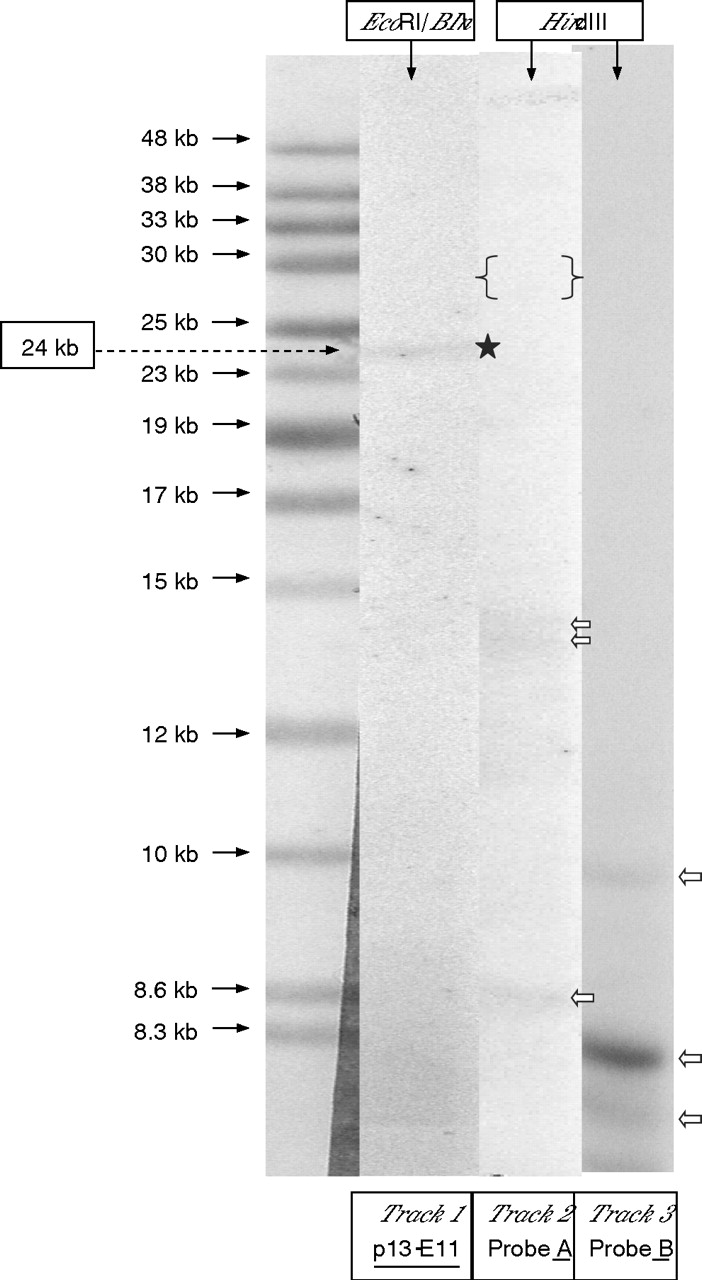
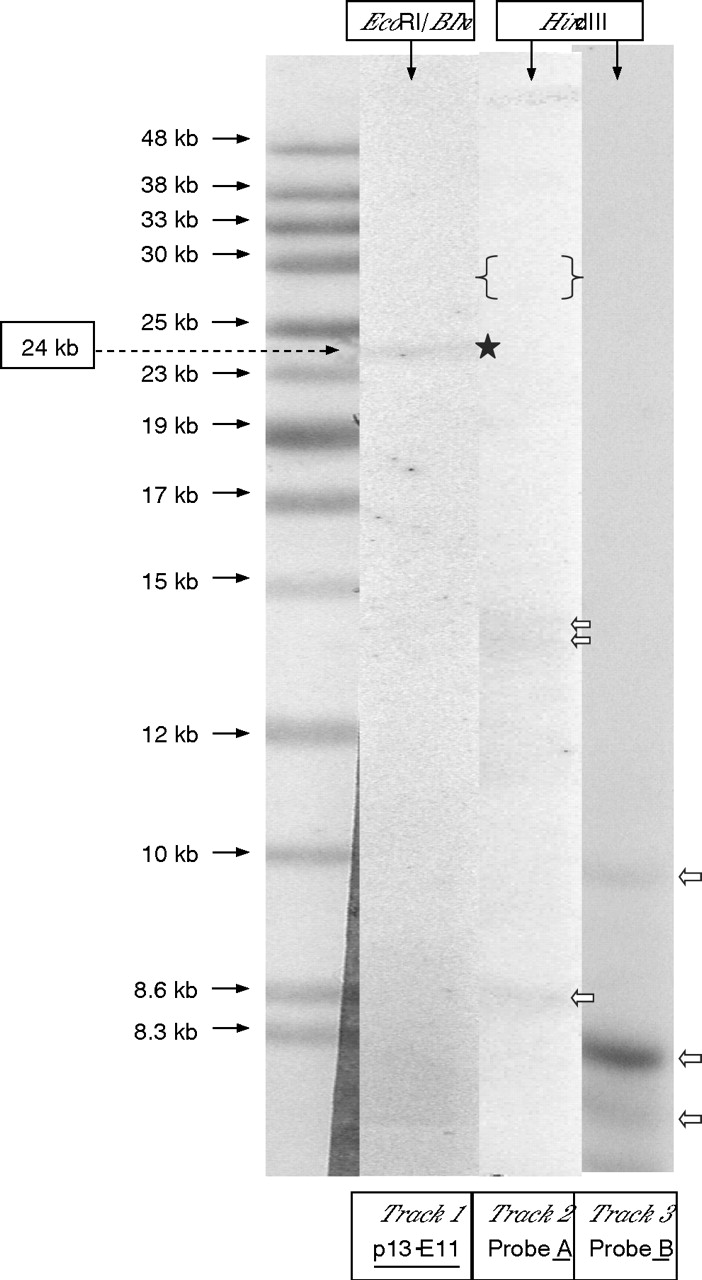
Figure 2 shows an example of the hybridisation results for probe p13-E11 to EcoRI and EcoRI/BlnI-digested DNA and probes p4qA and p4qB to HindIII-digested DNA from a typical patient with FSHD. PFGE-based analysis of 65 unrelated patients with FSHD, 15 from Turkey and 50 from the UK, showed the complete concurrence of a small (⩽11 repeats) D4Z4 array located on 4qA-defined 4q35-located subtelomere in each patient. However, analysis of the normal chromosome 4 in each of these 65 patients showed an almost equal frequency for the 4qA (54.5%) and 4qB (45.5%) alleles. In the more extensive LGE-based analysis of HindIII-digested standard DNA from 99 unrelated patients with FSHD, it was found that 97 of these samples exhibited a small 4q35-located 4qA allele compatible with their small (∼38 kb, ⩽11 repeats), EcoRI/BlnI fragment. However, in the DNA from the two remaining patients with FSHD, neither probe 4qA nor probe 4qB hybridised to any small 4q35-specific DNA fragments, even though the expected number of non-specific gel bands were observed with both probes. Figure 3 shows the hybridisation results from applying probes p13-E11, p4qA and p4qB to EcoRI/BlnI-digested and HindIII-digested DNA from one of these patients with FSHD .

Pulsed-field gel electrophoresis analysis of patient with facioscapulohumeral muscular dystrophy (FSHD) DNA. EcoRI and EcoRI/BlnI DNA digests hybridised to p13-E11 (track 1 and 2). HindIII DNA digests hybridised to probe 4qA (track 3) and probe 4qB (track 4). Chromosome 4-specific bands (white stars) and chromosome 10-specific bands (black stars) are indicated. All non-specific hybridising bands are identified (white arrows). The HindIII-digested DNA hybridises only to probe 4qA (track 3) and not to probe 4qB, indicating that this patient with FSHD has an AA genotype at 4q35.
{kind=link}
{kind=link}
{kind=link}
Analysis of DNA from a patient with facioscapulohumeral muscular dystrophy lacking 4qA and 4qB alleles. Track 1: DNA from patient digested with EcoRI/BlnI hybridised with p13-E11, a 4q35-derived 24 kb band is indicated (black star). Tracks 2/3: Patient DNA digested with HindIII and hybridised to probe 4qA2 and probe 4qB.3 No band comparable to the 24 kb EcoRI/BlnI was found, an ∼30 kb 4qA band would be expected (within the region of the gel marked { }). Only non-specific hybridising bands are present (white arrows).
DISCUSSION
Assessment of 4qA and 4qB allele frequencies in a large cohort of 164 unrelated patients with FSHD, which included 15 unrelated Turkish patients, confirmed the almost exclusive association between FSHD disease-expression and the presence of small (∼38 kb, ⩽11 repeats) D4Z4 repeat arrays located on small 4qA-defined 4qter subtelomeres. The FSHD patient sample surveyed more than double the numbers reported in two previous similar studies.8,9 As none of the normal individuals analysed showed a small (⩽38 kb) 4q35-located D4Z4 repeat array, we were unable to corroborate whether any of these were associated with a small 4qB-defined 4qter subtelomere, and would therefore be considered to be non-disease-associated.10
The 4q35-located 4qA/4qB allele frequencies determined in the 15 normal Turkish DNA samples showed an equal prevalency for 4qA-defined and 4qB-defined 4qter subtelomeres, showing no apparent difference in the 4qA/4qB allele frequencies in this ethnic subpopulation.
An additional rare type of 4qter subtelomere was identified after the repeated failure of hybridisation of both probes 4qA and 4qB to any small 4q35-specific DNA fragment from two patients with FSHD, known to have small (24 and 21 kb) EcoRI/BlnI fragments. None of the normal individuals analysed gave evidence for an alternative 4qter subtelomere, although this has been reported.11 It is not known if there is any relationship between this rare 4qter subtype and FSHD disease expression.
This large FSHD patient study has corroborated and strengthened the evidence for the almost exclusive association between disease expression and the location of the FSHD locus on a small 4qA-defined 4qter subtelomere. Confirmation of this essentially complete allelic association greatly increases confidence in implementing the use of the 4qA and 4qB probes in routine molecular diagnoses of FSHD. This may be especially important in those FSHD cases which show extreme variability in their clinical presentation.
Acknowledgments
We thank all the clinicians who have provided us with FSHD patient samples, especially Dr Peter Lunt (Bristol), Dr Mark Rogers (Cardiff), and Dr Piraye Serdaroglu (Turkey). We gratefully acknowledge the financial support of the Association Française contre les Myopathies (AFM). Finally, we thank the many patients with FSHD, and their families, who have for many years given us their full support and encouragement.
REFERENCES
Footnotes
-
Published Online First 20 September 2006
-
Competing interests: None.