Article Text

Statistics from Altmetric.com
- HGPS, Hutchinson–Gilford progeria syndrome
- MAD, mandibuloacral dysplasia
- MADA, MAD with fat loss affecting only the extremities
- MADB, MAD with generalised fat loss
- RD, restrictive dermopathy
Hutchinson–Gilford progeria syndrome (HGPS; OMIM 176670) is an extremely rare but devastating disorder that mimics premature aging.1–3 Affected children appear normal at birth but typically develop failure to thrive in the first two years. Other features include alopecia, micrognathia, loss of subcutaneous fat with prominent veins, abnormal dentition, sclerodermatous skin changes, and osteolysis of the clavicles and distal phalanges. The mean age of death is at age 13 years, most commonly due to atherosclerosis. HGPS is mainly sporadic in occurrence, but a genetic cause has now been implicated following the identification of de novo heterozygous mutations in the LMNA gene in the majority of HGPS patients.4,5 A single family showing autosomal recessive inheritance of homozygous LMNA mutations has also been reported.6
LMNA encodes lamins A and C, components of the nuclear lamina, a meshwork underlying the nuclear envelope that serves as a structural support and is also thought to contribute to chromatin organisation and the regulation of gene expression.7,8 Interestingly, mutations in LMNA have recently been associated with at least eight inherited disorders, known as laminopathies, with differential dystrophic effects on a variety of tissues including muscle, neurones, skin, bone, and adipose tissue (reviewed in Mounkes et al9). However, the realisation that these disorders share common genetic defects has led to clinical re-evaluation, with emerging evidence of significant phenotypic overlap.10 Hence the laminopathies might reasonably be considered as a spectrum of related diseases.
HGPS has phenotypic similarities to several other laminopathies, in particular the atypical Werner’s syndrome11 and mandibuloacral dysplasia (MAD; OMIM 248370 and 608612).12 These diseases are associated with lipodystrophy,3,13 which is the most prominent feature of another laminopathy, familial partial lipodystrophy of the Dunnigan variety (OMIM 151660).14 MAD has been further classified as two types according to whether the fat loss affects only the extremities (MADA) or is generalised (MADB).13 Patients with classical HGPS, caused by heterozygous mutation of the LMNA gene, appear to show some skeletal anomalies typical of MAD, including micrognathia and osteolysis of the distal phalanges and clavicles. However, a kindred with an atypical form of HGPS, with homozygous LMNA mutations revealed the more prominent radiological features of MAD, including acro-osteolysis and hypoplasia or absence of the clavicles.6,15
In addition to LMNA, MADB has been associated with compound heterozygous mutation of the ZMPSTE24 gene,16 which encodes a zinc metalloproteinase necessary for the proteolytic processing of prelamin A to form mature lamin A. This involves farnesylation of the cysteine residue within the C-terminal CAAX motif of prelamin A, followed by removal of the AAX by ZMPSTE24. The cysteine residue is then methylated and a final proteolytic cleavage, now also thought to be performed by ZMPSTE24,17 removes a further 15 C-terminal residues to produce mature lamin A. In agreement with this, Zmpste24 knock-out mice are unable to process prelamin A and have a phenotype reminiscent of human laminopathies.18,19 Recently, mutations in LMNA and ZMPSTE24 were also found to be associated with restrictive dermopathy (RD; OMIM 275210), a neonatal disorder involving tight adherent skin, joint contractures and respiratory insufficiency, together with features of the progeroid syndromes.20 A single ZMPSTE24 mutation, c.1085_1086insT, previously reported in MAD,16 was identified in seven of the nine individuals studied. However, in four of these seven cases, the mutation was inherited from one unaffected parent. The second mutation was undetermined in these patients. It is therefore unclear whether the ZMPSTE24 gene alone is responsible for restrictive dermopathy or acts in concert with other defective gene(s).
Key points
-
Hutchinson–Gilford progeria syndrome (HGPS) is a rare but devastating genetic disorder that mimics premature aging. Most cases are caused by heterozygous mutations in LMNA, encoding lamin A/C. The allelic disorder, mandibuloacral dysplasia (MAD), shares many features with HGPS and can also result from homozygous mutations in ZMPSTE24, which encodes the enzyme responsible for proteolytic processing of prelamin A. Heterozygous mutation of ZMPSTE24 was recently found to be associated with restrictive dermopathy (RD), a lethal neonatal disorder characterised by tight skin and sharing features of MAD/HGPS.
-
Compound heterozygous ZMPSTE24 mutations, c.1085_1086insT (Leu362PhefsX19) and c.794 A→G (N265S), were identified in a two year old girl with a severe early onset progeroid phenotype displaying features of HGPS, MAD, and RD.
-
Western blot analysis of skin fibroblasts showed defective lamin A processing, resulting in reduced levels of mature lamin A and increased levels of the prelamin A precursor.
-
Primary skin fibroblasts showed abnormal nuclear morphology and these abnormalities increased in severity upon culturing.
-
Our data widen the spectrum of progeroid phenotypes associated with ZMPSTE24 mutation and for the first time provide evidence that these mutations lead to defective prelamin A processing and abnormal nuclear morphology in humans.
Here, we report the identification of compound heterozygous ZMPSTE24 mutations in a two year old girl with a severe progeroid phenotype showing features of HGPS, MAD, and RD. Furthermore, we demonstrate defective prelamin A proteolytic processing and severe abnormalities in nuclear morphology in this patient.
METHODS
Subjects and samples
Genomic DNA samples were obtained from peripheral blood lymphocytes of patient FT and her parents, following ethical approval and informed consent. FT fibroblasts were obtained by skin biopsy at age four months and control fibroblasts were kindly provided by S Brown (Hammersmith Hospital, London). Written consent for publication of the patient images was obtained from the parents of FT and is available upon request.
Polymerase chain reaction and sequence analysis
Analysis of the LMNA and ZMPSTE24 genes was carried out as described previously.14,16 ZMPSTE24 cDNA, accession number NP_005857.2, was used for naming of mutations.
794 A→G allele genotyping
We analysed genomic DNA from 50 unrelated individuals using the allele discrimination assay from ABI for 794 A→G genotyping (Assays-by-Design Service—SNP genotyping, Applied Biosystems, Foster City, California, USA). The genomic DNA was amplified with optimised primers and probes, as suggested by the manufacturer. This assay is based on the binding of fluorescently labelled probes to the amplified products. The release of fluorescently labelled probes was then read by 7700 sequence detection system (ABI). We further amplified seven genomic DNAs with polymerase chain reaction (PCR) primers for exon 7 and directly sequenced the PCR product as described for mutational analysis.
Cell culture
Fibroblasts were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 4500 mg/l glucose supplemented with 10% fetal bovine serum and 100 U/ml penicillin-streptomycin and were maintained at 37°C, 5% CO2. Upon reaching confluence, the cells were trypsinised and subcultured. FT and control fibroblasts were maintained at equivalent passage numbers for each experiment, which ranged from 18 to 21.
Western blotting of cell extracts
Total cell extracts were prepared from cells grown to confluence in 80 cm2 flasks. Following trypsinisation, cell pellets were resuspended in phosphate buffered saline (PBS), boiled in an equal volume of 2× Laemmli buffer, then separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Gels were transferred to nitrocellulose membranes (Schleicher & Schuell Bioscience, Keene, New Hampshire, USA) using a Hoefer SemiPhor semi-dry transfer unit. Membranes were incubated with primary antibodies: lamin A/C (JOL2; Chemicon International, Temecula, California, USA), prelamin A (SC-6214; Santa Cruz Biotechnology, Santa Cruz, California, USA), or α-tubulin (DM 1A; Sigma, St Louis, Missouri, USA). Following extensive washing, the membranes were then incubated with peroxidase conjugated anti-mouse or anti-goat secondary antibodies (Sigma) and proteins revealed using the ECL plus western blotting detection system (Amersham Biosciences, Amersham, UK).
Indirect immunofluorescence and electron microscopy
For immunofluorescence studies, cells grown on glass coverslips were fixed in methanol at −20°C. Primary antibodies were as for western blotting; secondary antibodies used were rabbit anti-mouse AlexaFluor 488 and donkey anti-goat AlexaFluor 594 (Molecular Probes, Eugene, Oregon, USA). Antibodies were applied in PBS/3% bovine serum albumin (BSA) for one hour at room temperature. DNA was stained with 0.2 μg/ml Hoechst 33258 (Sigma). Coverslips were then mounted in 80% glycerol/3% n-propyl gallate (in PBS) and viewed with a Nikon TE300 inverted microscope using an ORCA ER charge couple device camera (Hamamatsu) and Openlab 3.09 software (Improvision). Nuclear morphology was observed in at least 300 cells per coverslip and multinucleate cells or those that possessed more than two lobulations were classed as abnormal.
For electron microscopy, a skin biopsy was fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2, and postfixed in 1% osmium tetroxide. The sample was processed and embedded in Agar 100 resin. Ultrathin sections were cut and stained with uranyl acetate in 70% alcohol and Reynolds lead citrate before examination using a Joel 100 CX transmission electron microscope.
RESULTS
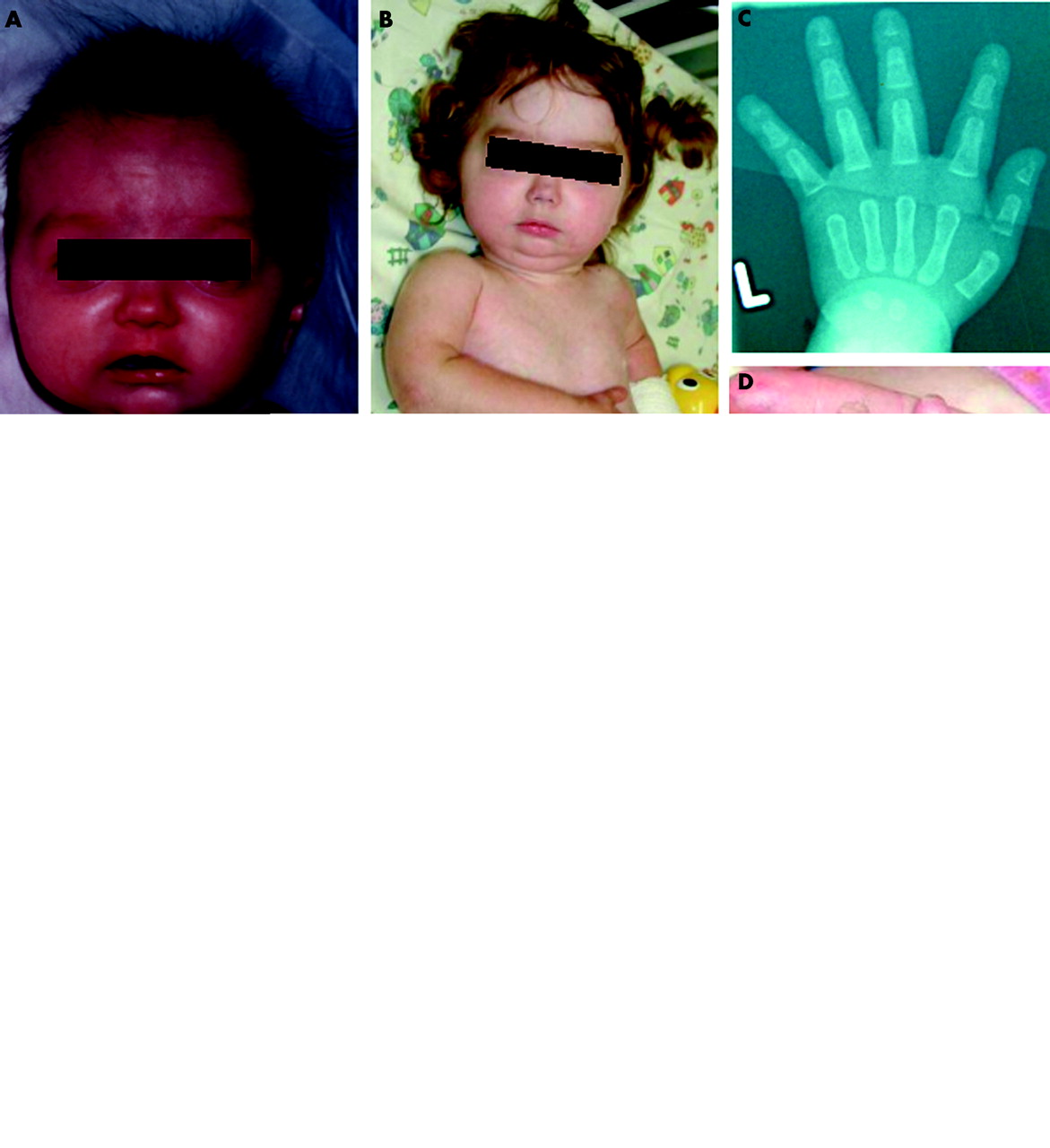
Clinical features (fig 1)

Clinical features observed in patient FT. (A) Age 4 months, showing her fine hair, “glyphic” nose, and micrognathia. (B) Age 2 years 4 months. (C–D) Radiology at age 4 months showing acro-osteolysis (C) and photograph at age 2 years 4 months (D) showing short, bulbous distal phalanges and nails. (E) Chest x ray age 2 years 4 months, showing marked osteolysis of the clavicles with an irregular mass of callus overlying the left clavicular remnant (arrow) and a fracture of the left upper humerus (arrowhead). (F) Dry, shiny and tight skin on lower leg at 4 months. Written consent for publication of these images was obtained from the parents of FT.
FT was the second child born to healthy, unrelated parents. Her sister, now aged eight years, remains healthy. The pregnancy was complicated by severe hyperemesis and ended with a spontaneous vaginal delivery at 32 weeks. Neonatal alloimmune thrombocytopenia required treatment with platelet transfusions. At 10 weeks, examination revealed restricted extension at the knees and bowing of the legs, dry shiny and tight skin over the abdomen, trunk, and lower limbs, prominence of the calf muscles because of lipoatrophy, mottling of the skin of the hands and arms, clenched fists, plagiocephaly, and craniotabes. Further evaluation revealed a bright cheerful responsive infant with fine hair, wide anterior and posterior fontanelles with palpable Wormian bones and wide lambdoid sutures, a small mandible with two erupting tooth buds, short distal phalanges and nails. The skin and subcutaneous tissues over the abdomen had a woody feel. The skin of the lower legs was firmly adherent to the underlying muscle and subcutaneous fat was absent. Movements were restricted at the hips, knees, and ankles.
Radiology revealed a thin skull with extensive Wormian bones; a small mandible; acro-osteolysis; marked osteolysis of both clavicles; and lytic lesions in the right upper tibia, left upper humerus, and cervical spine. She had continuing problems with poor weight gain, upper airway obstruction, dry itchy skin, cutaneous and subcutaneous oedema, transient ischaemic attacks, a non-healing fracture of the left clavicle, which became surrounded by extensive callus, and a fracture of the left upper humerus.
Her developmental progress remained normal although walking was restricted by stiffness of the skin and joints. Eventually, areas of the skin became extremely sclerotic (morphoea-like) and large areas broke down and became infected. She developed respiratory failure and died at 2 years 9 months. Table 1 compares the features observed in FT with those typical of HGPS, MAD, and RD.
Clinical features of early onset progeroid syndromes
Mutation analysis
Direct sequence analysis of genomic DNA from FT revealed no mutation in the exons or intron/exon boundaries of the LMNA gene (data not shown). Our patient shared several features with those reported earlier in a patient with ZMPSTE24 mutations.16,23 We therefore screened for mutations in this gene and identified compound heterozygous mutations. The first mutation, c.1085_1086insT in exon 9, has been reported previously in a patient with MADB16 and results in a frameshift and premature protein truncation Phe361fsX379 (Leu362PhefsX19, according to standard mutation nomenclature guidelines at http://www.genomic.unimelb.edu.au/mdi/mutnomen). The second mutation, c.794 A→G in exon 7, results in substitution of asparagine with serine at amino acid 265 (N265S). This mutation was not observed in 100 control chromosomes. The parents were each heterozygous for one of the two mutations, yet had no obvious phenotype, observations that support an autosomal recessive inheritance for the severe progeroid phenotype segregating within this family.
Defects in prelamin A processing
To determine whether the identified ZMPSTE24 mutations have any impact on prelamin A processing, we carried out western blotting on cultured skin fibroblasts. Probing of total cell extracts with antibodies against lamin A/C showed an additional higher molecular weight band in the patient sample, consistent with the size of prelamin A (fig 2, upper panel). The levels of mature lamin A were significantly reduced in FT, whereas lamin C levels were unchanged compared with the control. The higher molecular weight band was confirmed as prelamin A by reprobing the western blot with anti-prelamin A antibodies (fig 2, middle panel). A longer exposure revealed a faint prelamin A band in the control sample, showing that prelamin A is expressed at low levels in control cells (data not shown).

Western blot analysis of WT and FT skin fibroblasts. Whole cell extracts from cultured fibroblasts were western blotted with antibodies against lamin A/C, prelamin A, or α-tubulin (as a loading control). In the upper panel, the WT lamin A and lamin C bands are indicated. Note the presence of an additional band in FT (arrow).
Indirect immunofluorescence microscopy revealed normal localisation of lamin A/C to the nuclear periphery in the patient fibroblasts (fig 3A). Strikingly, however, fibroblasts from FT showed a high degree of abnormal nuclear morphology compared with the mainly uniform oval or round nuclei of wild-type cells. Many nuclei were lobulated or multilobulated and some cells appeared multinucleate. In both WT and patient fibroblasts, there was an increase in the percentage of abnormal nuclei and the severity of the abnormalities at higher passage numbers. Unfortunately, patient cells below passage number 17 were not available and at the higher passages studied, even WT cells began to display significant abnormalities. However, the nuclear defects were always more numerous and more severe in the patient cells. At passage number 18, 7.5% of WT nuclei and 41% of FT nuclei showed significant abnormalities, and this increased to 56% and 77%, respectively, by passage 21. Both cell lines entered senescence at passage 22. Immunodetection with prelamin A antibodies confirmed the presence of increased levels of prelamin A in FT fibroblasts (fig 3B). Prelamin A co-localised with lamin A/C at the nuclear periphery, indicating that unprocessed lamin A is able to polymerise and incorporate into the nuclear lamina. Other nuclear envelope proteins—including emerin, nucleoporins, and lamin B—were correctly localised in all nuclei (data not shown).

{kind=link}
{kind=link}
{kind=link}
Microscopic analysis of WT and FT skin fibroblasts. (A, B) Immunofluorescence staining of cultured fibroblasts with lamin A/C (green) and prelamin A (red) antibodies. In (B), green and red channels are merged and DNA, stained with DAPI, is in blue. (C) Electronmicrograph of a typical nucleus from freshly biopsied FT skin fibroblasts. Red arrows indicate nuclear envelope invaginations. Scale, 16 000× magnification.
While we had access only to relatively high passage number cultured patient fibroblasts, electron microscopy had been performed at the time on the freshly biopsied skin fibroblasts. Examination of these samples revealed that, although internal nuclear organisation was unperturbed, the nuclear envelope was already irregular in shape and forming lobulations (fig 3C).
Thus our data show that excessive prelamin A expression at the nuclear lamina disturbs nuclear structure and leads to the development of a severe, early onset progeroid phenotype.
DISCUSSION
This report identifies ZMPSTE24, in addition to LMNA, as a causative gene for a severe progeroid disorder resembling HGPS. Clearly, our finding has important implications for genetic counselling, as the autosomal recessive inheritance identified in this kindred is associated with a 25% recurrence risk, in contrast to the low (<3%) recurrence risk described in classic HGPS, the majority of which arise from de novo LMNA mutations. While a clinical diagnosis of HGPS was established in FT, she appeared to share features also seen in restrictive dermopathy, particularly the presence of tight skin susceptible to oedema and restricted joint extension, and features which are common to HGPS and MAD, including clavicular and mandibular hypoplasia and osteolysis of the distal phalanges and clavicles. Thus establishing the molecular basis of the progeroid disorder serves to extend the apparent phenotypic overlap between the laminopathies.
The c.1085_1086insT ZMPSTE24 mutation we identified was previously described in a patient with MAD16 and predicts premature truncation at amino acid 380 of the 475 residue protein. On the other hand, the N265S missense mutation has not previously been reported. Interestingly, the MAD patient described previously harboured a different missense mutation (W340R) on the second allele.16 This patient presented at age 2 and survived to 24 years. The earlier onset and increased severity of the disease in our patient may at least in part be attributable to functional differences between these alleles. That this child had a more severe form of HGPS than LMNA associated forms correlates with our finding that freshly biopsied skin fibroblasts already showed nuclear deformation. In cultured fibroblasts from classic HGPS patients carrying the common LMNA c.1824C→T mutation, nuclear morphology is normal at least up to a passage number of 6.24
The c.1085_1086insT mutation has also been reported in seven individuals with restrictive dermopathy.20 As this mutation was present in an unaffected parent in four cases, it is clearly insufficient by itself to cause disease. The nature of the additional mutation(s) is unknown but its identification may explain the extremely severe phenotype seen in these patients.
Given the known role of ZMPSTE24 in prelamin A processing, the ZMPSTE24 mutations identified would be predicted to disrupt this process, leading to increased prelamin A and reduced or absent mature lamin A. This was indeed observed in cell extracts from cultured fibroblasts. Unlike Zmpste24 knock-out mice, in which mature lamin A is completely absent,18,19 some processing of prelamin A did occur in patient fibroblasts, as mature lamin A was produced. This suggests that some active ZMPSTE24 is produced, although this is not sufficient for complete processing of prelamin A. The Leu362PhefsX19 mutant has previously been shown to be functionally inactive in a yeast halo assay testing its ability to complement a processing defect in the STE24 homologue.16 Thus the observed partial processing of prelamin A is likely to be caused by the N265S missense allele retaining residual proteolytic activity (Agarwal AK, Garg A, manuscript submitted). In contrast, lamin C does not undergo proteolytic processing and hence is unaffected by ZMPSTE24 mutation.
We observed severe abnormalities in nuclear morphology in the patient fibroblasts compared to wild-type cells. Wild-type prelamin A accumulation also correlates with increased nuclear deformation in cells from HGPS patients expressing a truncated lamin A, termed “progerin”, caused by the common c.1824C→T LMNA mutation.24 In addition, individuals heterozygous for a null LMNA allele, which reduces the expression of both lamin A and lamin C, present with a muscular dystrophy phenotype.25 Taken together, this suggests that it is the presence of excess prelamin A that has a dominant negative effect on nuclear envelope structure. As low levels of prelamin A were detected in WT cells, it is likely that a critical level of prelamin A must be reached to trigger the defects seen in progeroid disorders. One explanation is that excessive levels of prelamin A may disrupt lamina polymerisation or interactions with other nuclear proteins, leading to destabilisation of the nuclear envelope. While we did not detect any change in the localisation of emerin, nucleoporins, or lamin B, A-type lamins are known to interact with a wide range of other nuclear proteins. Recent proteomics studies also suggest that there are additional uncharacterised nuclear envelope proteins, many of which are likely to interact with lamin A.26
Conclusions
Our results are critical for the provision of detailed genetic counselling because they clarify the range of molecular defects underlying these disorders. Furthermore, we have shown that ZMPSTE24 mutations in humans lead to abnormal prelamin A processing, lending support to the importance of lamin A processing in the aetiology of progerias.
Note added in proof
Fong et al have recently demonstrated that the progeria-like phenotype of Zmpste24 knockout mice can be alleviated by reducing WT lamin A expression when crossed to a heterozygous Lmna knockout background.27 These results further support our conclusion that excess prelamin A has deleterious effects on the cell.
Acknowledgments
We would like to thank G Anderson and M Malone (Great Ormond Street Hospital, London) for performing electron microscopy, S Brown (Hammersmith Hospital, London) for providing control skin fibroblasts, F Khawaja for assistance with LMNA sequencing and M Millay for ZMPSTE24 sequencing. We also acknowledge the late R Winter for his initial clinical assessment. This research was funded by a BHF project grant to RCT and National Institutes of Health grant R01-DK54387 to AG. DS is supported by an MRC PhD studentship.
REFERENCES
Footnotes
-
Competing interests: none declared