Article Text

Abstract
Objective: To analyse somatic molecular changes, clinicopathological features, family history, and germline mutations in families with colorectal cancer (CRC).
Methods: Molecular changes (K-ras and β-catenin mutations, chromosome 18q allele loss (LOH), APC LOH, microsatellite instability (MSI), and expression of β-catenin and p53) were examined in four series of CRC patients with proven or probable hereditary disease: hereditary non-polyposis colon cancer (HNPCC); MYH associated polyposis (MAP); multiple (>5) colorectal adenomas without familial adenomatous polyposis (FAP); and other families/cases referred to family cancer clinics (FCC series). HNPCC was diagnosed using a combination of germline mutation screening and tumour studies. A series of unselected CRC patients was also studied.
Results: There was overlap between genetic pathways followed by each type of CRC, but significant differences included: increased frequency of K-ras mutation and reduced frequency of APC LOH in cancers from MAP, but not from multiple adenoma patients; reduced frequency of LOH in HNPCC CRCs; and increased MSI in CRCs from HNPCC, but not from FCC or multiple adenoma patients. HNPCC was apparently detected efficiently by combined germline and somatic analysis. Cancers from the FCC, unselected, and multiple adenoma series shared similar molecular characteristics. In the FCC and multiple adenoma series, hierarchical cluster analysis using the molecular features of the cancers consistently identified two distinct groups, distinguished by presence or absence of K-ras mutation.
Conclusions: While K-ras mutation status is known to differentiate hereditary bowel cancer syndromes such as MAP and FAP, it may also distinguish groups of non-HNPCC, FCC patients whose disease has different, as yet unknown, genetic origins.
- AFAP, attenuated familial adenomatous polyposis
- BER, base excision repair
- CRC, colorectal cancer
- FAP, familial adenomatous polyposis
- HNPCC, hereditary non-polyposis colon cancer
- LOH, loss of heterozygosity
- MAP, MYH associated polyposis
- MSI, microsatellite instability
- colorectal cancer
- hereditary non-polyposis colon cancer
- K-ras
- molecular pathway
Statistics from Altmetric.com
- AFAP, attenuated familial adenomatous polyposis
- BER, base excision repair
- CRC, colorectal cancer
- FAP, familial adenomatous polyposis
- HNPCC, hereditary non-polyposis colon cancer
- LOH, loss of heterozygosity
- MAP, MYH associated polyposis
- MSI, microsatellite instability
About one third of all colorectal cancers (CRCs) have some inherited basis.1 About 5% of all CRCs arise as part of the known Mendelian syndromes, principally hereditary non-polyposis colon cancer (HNPCC), familial adenomatous polyposis (FAP), and MYH (MUTYH) associated polyposis (MAP) (reviewed by Kemp et al2). Classically, “sporadic” colorectal adenomas arise following two APC mutations, with carcinoma occurring after the adenoma has progressively acquired mutation in K-ras, loss of chromosome 18q, and mutation or overexpression of p53.3 Some sporadic CRCs develop along a different pathway in which BRAF rather than K-ras mutations occur, there is a lower frequency of p53 mutations and a higher frequency of BAX mutations, the karyotype is near diploid, and deficient DNA mismatch repair renders the tumour microsatellite unstable (MSI+) (reviewed by Jass et al4).
FAP tumours appear to develop along similar genetic pathways to sporadic CRCs, probably because biallelic APC mutations are the initiating events in both cases,5,6 although FAP tumours have certain distinguishing features, such as a low frequency of K-ras mutations.7 Tumorigenesis in HNPCC follows a pathway similar to MSI+ sporadic CRCs, although in some HNPCC cancers β-catenin mutations substitute for APC mutations, and HNPCC tumours generally have K-ras rather than BRAF mutations.8 MAP CRCs follow another distinct pathway, being near diploid and microsatellite stable (MSI−), with a high frequency of APC mutations (but a low frequency of allelic loss), and a high frequency of K-ras mutation.9 In general, the genetic pathways of HNPCC and MAP carcinogenesis result from the underlying genetic instability specific to each condition, namely deficient DNA mismatch repair (MMR) and base excision repair (BER), respectively.
The differences between the genetic pathways of carcinogenesis in FAP, HNPCC, MAP, and sporadic CRCs partly reflect three factors. First, owing to their DNA sequence, some genes are susceptible to certain types of mutation; thus the 10 base pair (bp) oligoadenine tract in TGFBR2 is prone to mismatch slippage in HNPCC10 and tends to occur in preference to the SMAD4/SMAD2 mutations seen in MSI-CRCs, even though the latter might provide a greater functional advantage.11 Second, there is probably selection against co-occurrence of certain changes in CRCs—for example, a high frequency of allelic loss or aneuploidy/polyploidy rarely occurs together with defective MMR or BER.12 Third, some changes (for example, the missense β-catenin mutations in HNPCC) are probably coselected with other mutations which have been driven by the underlying genetic instability.13
The 25–30% of CRCs with an inherited basis, but outside the known Mendelian syndromes, probably result from unknown moderate penetrance or low penetrance predisposition alleles. Very little is known about the somatic genetic pathways followed by familial CRCs outside HNPCC, MAP, and FAP. In a large unselected series of CRCs, Slattery et al14 found no association between family history and K-ras mutation or MSI status, but did find a weak association between p53 mutation and a positive family history. Abdel Rahman et al15 analysed familial CRCs and found that nuclear β-catenin expression was more common in cancers from patients with identified germline mismatch repair mutations. In the mutation negative group, p53 mutation was associated with nuclear β-catenin expression.
If some of the inherited CRCs without a known genetic basis arose as a result of defective DNA repair, it would be expected—by comparison with HNPCC and MAP—that they would tend to have ploidy, mutation spectra, and frequencies of allelic loss that reflected this fact. It would be predicted, for example, that in DNA repair deficient CRCs, allelic loss at SMAD4 on 18q and at APC would be reduced in frequency, β-catenin and K-ras mutations might be more frequent, p53 mutations would be less frequent, and CRCs would tend to be near diploid. If, however, some of the remaining inherited CRCs resulted from moderate penetrance mutations in genes more similar to APC, genetic pathways of carcinogenesis would be similar to the sporadic, unselected CRCs; it is likely also that CRCs resulting from genes with low penetrance (or CRCs which were present in chance familial clusters) would follow genetic pathways that could not readily be distinguished from unselected cases. It might be possible, moreover, to distinguish different groups of CRC families of unknown genetic origin, based on differences between the clinicopathological or molecular features of their tumours.
In this study, we analysed CRCs from five series of patients and families: (1) HNPCC; (2) MAP; (3) patients with multiple colorectal adenomas, but without MAP or attenuated or classical FAP; (4) patients presenting to a family cancer clinic (FCC) with evidence of inherited disease, but without a molecular diagnosis of HNPCC, MAP, or FAP (FCC patients); and (5) a consecutive, unselected series of CRC cases with no information about their family history. We have screened the CRCs for mutations in K-ras and β-catenin, for MSI, for allelic loss at APC and on chromosome 18q (close to the SMAD4, SMAD2, and DCC loci), and for the expression of β-catenin and p53 proteins. We have compared the molecular findings with clinical, pedigree, and pathological data, and tested for evidence that the FCC cases can be clustered on the basis of molecular features into more than one group.
METHODS
Patient ascertainment
CRC cases were derived from patients and families referred to the Cancer Research UK family cancer clinic, St Mark’s Hospital, and the family cancer clinic, Guy’s Hospital. National research ethics guidelines were followed. Various families were incorporated into the study,16 using inclusion criteria similar to the Bethesda guidelines,17,18 as follows:
-
Amsterdam or modified Amsterdam criteria;
-
Amsterdam or modified Amsterdam criteria in all respects except no cancer occurring in an individual under 50 years;
-
one or more individuals diagnosed with CRC under 45 years;
-
one or more individuals with two HNPCC cancers (bowel or endometrial);
-
>5 adenomas to date in any individual (with fewer than 100 adenomas in total and excluding germline APC mutations);
-
three or more individuals affected by CRC in one generation.
Families from whom archival tumour tissue specimens could not be retrieved were excluded, although those without a living affected person were not automatically excluded. Pedigree information was obtained from family members and the presence of cancer confirmed, wherever possible, from hospital records, pathology reports, cancer registry records, or death certificates.
Clinical variables
Every included family was given a score in each of the following binary categories: the apparent mode of inheritance—dominant (multigenerational) or recessive (isolated case or single generation); the presence of one or more persons with two primary tumours (colorectal and/or endometrial cancer) in the family; the presence or absence of a woman with endometrial cancer in the family; the mean age at diagnosis of CRC (⩽45 or >45 years); the age at diagnosis of the youngest person with CRC or endometrial cancer (⩽45 or >45 years); predominance of right or left sided colorectal cancers in the family; and the presence or absence of a patient with five or more adenomas in the family. In addition, the number of persons affected by CRC in the family and the total number of other cancers (apart from colorectal or endometrial) developed by family members were noted. Table 1 summarises the clinical features of each family type.
The clinical features of the families and cases studied
Collection of blood and tumour samples from families
Living, affected family members were asked to give a 10 ml blood sample and consent for access to archival tumour tissue. Where affected family members were deceased, their next of kin was asked to give consent for access to archival tumour and normal tissue specimens. All available colorectal carcinomas were obtained from affected family members. The histological features of the tumours were taken from pathology reports or assessed by one of us (ATES) where no reports were provided. Microdissection of neoplastic and normal areas from paraffin embedded archival tissue was guided by reference to haematoxylin and eosin stained sections; tumour and normal DNAs were then extracted using a simple proteinase K digestion.
Germline mutation detection and family classification
From the 259 eligible families ascertained from the family cancer clinics of St Mark’s and Guy’s Hospitals, we subdivided cases (table 2) into those with germline mismatch repair mutations (HNPCC), biallelic MYH mutations (MAP), APC mutations (AFAP), or no mutation detectable in these genes. Molecular diagnosis of HNPCC was undertaken for all families and cases using the criteria of Lipton et al,16 which are based on a combination of mutation screening (MLH1, MSH2, and MSH6) and tumour analysis. For the latter, we used a combination of MSI testing and immunohistochemistry for MLH1, MSH2, and MSH6.16 For MSI testing, we used mononucleotide markers BAT26 and TGFBRII, and dinucleotide markers D5S346, D18S487, and D18S46, with bandshifts at two or more markers or at BAT26 alone being used to classify a tumour as MSI+.16 Forty three families were found to have HNPCC, of which only three contained individuals with five or more adenomas. MYH screening of all cases with three or more colorectal adenomas was undertaken as described,19 and 17 carriers of pathogenic biallelic MYH mutations were found. For APC screening of all cases with three or more colorectal adenomas we used a fluorescence-SSCP method adapted from described protocols,20 although no case was actually found to be APC mutant. The remaining families or cases (without HNPCC, MYH, or APC mutations) were subdivided into 49 “multiple adenoma” pedigrees (at least one individual with five or more proven colorectal adenomas to date) and 150 FCC families or cases (all others).
Numbers of families/cases studied from each of the five series and the frequency of each molecular change
Unselected series of colorectal carcinomas
An unselected series of 100 fresh frozen CRCs and paired normal bowel was obtained from St Mark’s Hospital, London; fixed tissue was obtained from the same tumours. All cancers contained more than 60% neoplastic cells, as assessed using routine histology. Clinicopathological data were obtained from hospital records. DNA was extracted from each tumour sample and paired normal bowel using standard methods. These samples were studied on an anonymised basis according to local research ethics guidelines and their features will be reported by Rowan et al (in preparation).
Immunohistochemistry for β-catenin and p53
Available colorectal cancers were tested by immunohistochemistry for overexpression of β-catenin and p53. Tumour sections (5 μm thickness) were analysed using the β-catenin mouse monoclonal antibody (Sc 7963) from Santa Cruz Biotechnology (Santa Cruz, California, USA) and p53 mouse monoclonal (M 7001) from Dako antibodies (Cambridge, UK), respectively, at 1/100 dilution after pressure cooking the sections for four minutes. After counterstaining with haematoxylin, the slides were examined by three independent observers (LRL, VJ, ATES). For β-catenin, nuclear expression was scored as positive if any nuclear staining and cytoplasmic staining was observed, and negative if membrane or cytoplasmic staining, or both, was seen. For p53, expression was scored as positive if more than 5% of tumour cells had nuclear staining and negative otherwise. For tumours without normal tissue, sections containing some normal tissue were used to provide an internal control in the case of β-catenin. For p53 controls, slides known to be positive for nuclear staining were used.
Mutation screening for K-ras and β-catenin
Mutations in K-ras (codons 12, 13, and 61) and β-catenin (exon 3) were detected in each cancer using direct sequencing in forward and reverse orientations as previously described.9
Allelic loss (LOH) analysis
LOH was assessed at the APC and SMAD4 loci (the latter of these also referred to as 18q LOH). Microsatellites very close to each locus ((D5S346 and D5S421 for APC; D17S487, D18S46, D18S474, and D18S35 for SMAD4) were typed in each cancer and in a sample of constitutional DNA. Constitutionally homozygous markers or markers showing MSI in tumours were scored as non-informative. Otherwise, at each marker, LOH was considered to be present if the area under one allelic peak in the tumour was less than 0.5 times or greater than 2 times that of the other allele, after correcting for the relative allelic areas using the constitutional DNA. If there was any discordance among markers, the markers closest to the gene of interest were given precedence in classifying the cancer.
Data scoring
In order to make analysis practicable, we scored each of the molecular variables as a single binary data point for each family or isolated case. If it was only possible to analyse a single cancer for that family, those results were used. However, we reasoned that where multiple cancers were available, we should obtain as much data as possible in order to reduce the effects of chance variation in genetic pathways, and then assess each family or case on the basis of all available cancers. For families or cases in which more than one cancer was analysed, we therefore scored molecular changes according to the most frequent result; for example, if two of three cancers had a particular change, that family was scored as positive for that change, but if one of three cancers had the change, the family was classed as negative. If the same number of cancers was with and without the change, we classed the family or case as positive.
Statistical analysis
Single variable tests (Fisher’s exact and Wilcoxon tests) and multivariable analysis (logistic regression) were undertaken using STATA 7.0. We carried out hierarchical cluster analysis using the cluster averagelinkage command of STATA. Occasional PCR failures or exhaustion of samples meant that not every data point was available for each of the tumours; missing data points for the cluster analysis were substituted by the mean across the whole patient set. The cluster generate command of STATA with two groups was used to partition out clustered groups of cases/families. Where testing specific hypotheses or confirming previously reported associations, or in the multivariable analysis, we used a threshold of p = 0.05 to indicate statistical significance. Where searching for new associations in the single variable analysis we used the more restrictive significance threshold of p = 0.01 in order to make some allowance for multiple testing.
RESULTS
We initially examined the frequency of each molecular change in the five series of patients or families (table 2). K-ras mutations were found at similar frequencies (0.27 to 0.34) in each of the series, except for the MAP patients (0.64); specifically, the K-ras mutation frequency in the carcinomas from MAP patients was significantly higher than in the multiple adenoma patients (p = 0.009, Fisher’s exact test) and higher than in all the other patient groups combined (p = 0.01, Fisher’s exact test). The frequency of LOH at APC ranged from zero in the MAP cancers and 0.36 in HNPCC to 0.58 in the multiple adenoma patients (table 2); with the clear exception of MAP cancers, which were significantly different from all other cancer types (details not shown), there were no statistical differences in APC LOH frequency among individual groups, although the difference was of borderline significance when comparing HNPCC and unselected tumours (frequency = 0.55; p = 0.066, Fisher’s exact test). The frequencies of 18q LOH ranged from 0.36 in HNPCC to 0.59 in the unselected group (table 2), the only significant difference being between these two groups (p = 0.037, Fisher’s exact test). As expected, MSI was present in all of the HNPCC cancers and none of the MAP cancers. About 10% of cancers from multiple adenoma patients, FCC patients, and unselected cases were MSI+. β-Catenin mutations were almost exclusively found in the HNPCC cancers, as previously reported,13 but nuclear expression of β-catenin protein was actually less frequent in these cancers than in others (although not significantly so). Overexpression of p53 was similarly frequent in all cancer types and no differences were observed (table 2). On multivariable analysis, no additional molecular discriminators between patient groups were found (details not shown).
We then focused on the FCC patient series. We carried out searches for pairwise associations between the molecular variables and for associations between the molecular and clinicopathological variables. The only association detected between the molecular variables was between LOH at APC and LOH on chromosome 18q; 30 of 42 CRCs with APC LOH also had loss on 18q, whereas only 15 of 38 tumours without APC LOH had loss on 18q (p = 0.004, Fisher’s exact test). This observation may reflect the common co-occurrence of these changes in aneuploid/polyploid lesions.12 Logistic regression analysis confirmed the association between APC LOH and 18q LOH, but did not reveal further associations between the molecular variables.
K-ras mutation, APC LOH, 18q LOH, MSI, and nuclear β-catenin expression were not associated with any of the clinicopathological variables in the FCC patient series. Absence of p53 overexpression was associated, with borderline significance, with a lower age of the youngest person in the family with colorectal or endometrial cancer (Fisher’s exact test, p = 0.019). For those families with p53 overexpression, the mean age of the youngest affected individual was 46 years (median = 46, interquartile range 39 to 52); for those without p53 overexpression, mean age of the youngest affected individual was 38 years (median = 40, interquartile range 30 to 43). Multivariable analysis did not reveal any additional associations between the molecular and clinicopathological data.
Cluster analysis was then used to suggest groups of FCC patients who might have similar clinical features and hence arise from similar genetic origins. The FCC patients were divided into four groups of between 30 and 40 at random. Hierarchical cluster analysis by clinicopathological features in each group revealed no consistent clusters (details not shown). Cluster analysis using the five molecular variables (K-ras mutation, APC and 18q LOH, β-catenin expression, and p53 expression) was then undertaken. Following clustering, for each of the four cluster replicates the families/cases were partitioned into two dissimilar groups, with the aim of identifying molecular factors which discriminated consistently between different types of patient (fig 1). In all the four cluster replicates, presence or absence of K-ras mutation was a highly consistent discriminant. Only five patients or families failed to cluster correctly by K-ras status. (A nominal test of the association between K-ras status and cluster group, excluding missing data points, showed a highly significant association in all four groups; p<0.002 in all cases by Fisher’s exact test.) None of the other molecular variables was discriminatory in more than one of the four replicates.

Results of hierarchical cluster analysis on FCC patients split into four sets. Each branch of the dendrogram represents one family or patient. The group (1 or 2) assigned by STATA cluster group command is shown below each branch of the dendrogram. Note that for all four dendrograms, the cancers of those patients or families in group 1 almost invariably had no K-ras mutation, and those in group 2 almost invariably had a mutation; in all, five discordant families (*) were found.
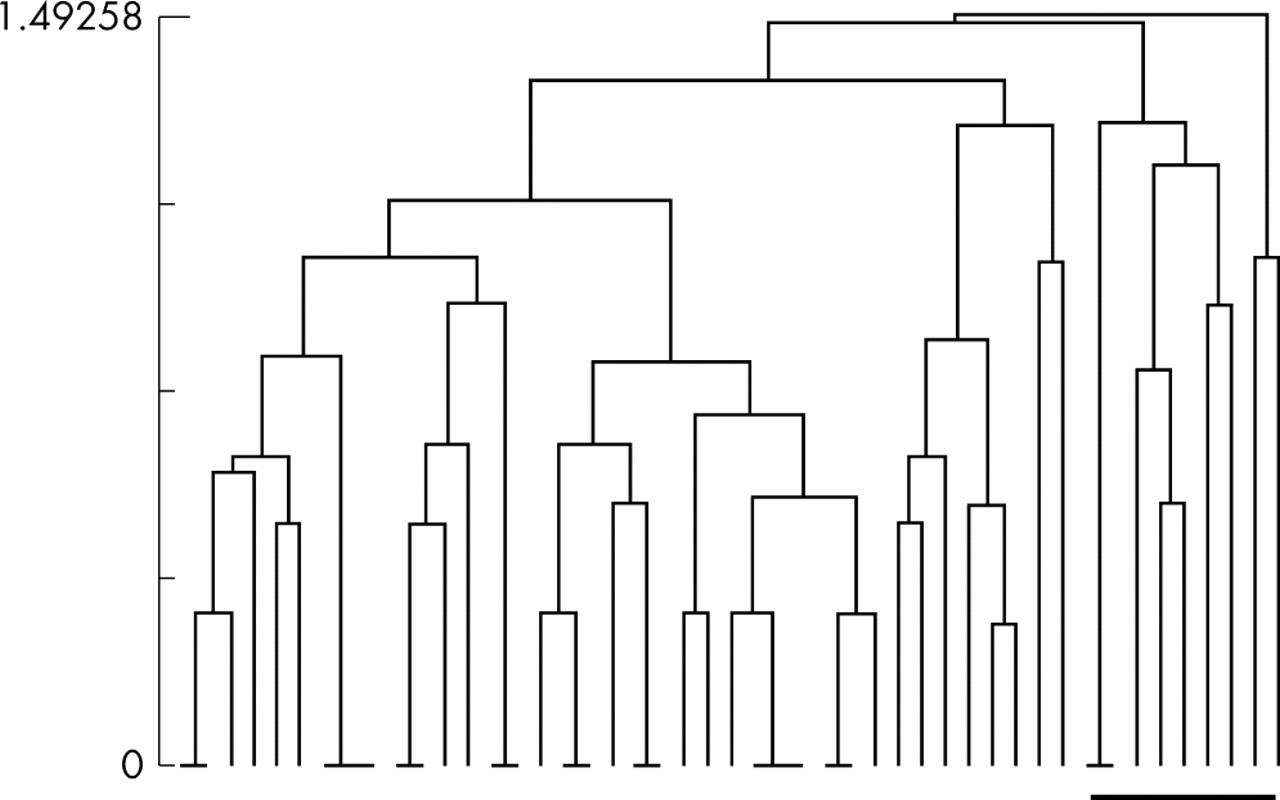
Finally, we analysed the multiple adenoma series. Molecular data generally supported those in the FCC series. In particular, LOH at APC and on chromosome 18q were more strongly associated overall (p = 0.001, Fisher’s exact test). Cluster analysis on the entire multiple adenoma series using the five molecular variables showed no consistent discriminator when the families or cases were partitioned into two dissimilar groups. However, on partitioning into three groups, presence or absence of K-ras mutation was a perfect discriminant, all nine tumours with K-ras mutations being in groups 2 and 3 and all non-mutant tumours being in group 1 (fig 2). None of the other molecular variables was discriminatory.

{kind=link}
{kind=link}
Results of hierarchical cluster analysis on multiple adenoma patients. Each branch of the dendrogram represents one family or patient. The bar indicates the K-ras mutant cancers, which are all clustered together.
DISCUSSION
It is well established that different germline susceptibilities to cancer determine, at least in part, the genetic pathways which those cancers follow and hence tumour morphology and behaviour. In this study, we extended this analysis to family cancer clinic patients without known Mendelian conditions, and compared our findings with tumours from HNPCC and MAP, from non-FAP patients with multiple adenomas, and from an unselected series of colorectal carcinomas. For many patient or family types, CRCs followed similar or overlapping genetic pathways, but we also found a small number of specific differences.
MAP cancers had molecular features that were different from those of the multiple adenoma patients. K-ras mutations were particularly frequent in MAP and, specifically, were more common than in the multiple adenoma cases. APC LOH was infrequent in MAP compared with other cancers, including those from multiple adenoma patients. MSI was absent in tumours from MAP patients, but present in multiple adenoma patients’ cancers at a frequency similar to the unselected series. Given that these data in MAP cancers result from the underlying defective BER in that condition21—and hence an increased rate of G→T mutations—our results suggest that multiple adenoma patients without germline mutations in MYH or APC do not result from a mutator phenotype at the base pair level which is comparable with that of MAP. Indeed, the molecular features of the cancers from the multiple adenoma patients were essentially indistinguishable from those of the FCC and unselected cancers.
As others have found,22 the frequencies of LOH at APC and on chromosome 18q were lower in HNPCC carcinomas than in the other series (although only significantly so in the latter case). These changes could not, however, be used to distinguish reliably between HNPCC patients from the FCC cases and the unselected series. The frequency of MSI was only slightly higher in the FCC patient series than in the unselected series and multiple adenoma cancers. The data therefore suggest that our use of mutation screening of MLH1, MSH2, and MSH6—coupled with immunohistochemistry and MSI analysis16—diagnoses the great majority of HNPCC cases. However, given that MSI frequency generally increases with patient age23 and that the FCC cases had a mean age of 51 compared with 67 for the unselected series, it would be expected that the frequency of MSI would be lower in the FCC than the unselected cancers. A few FCC cases are probably, therefore, true HNPCC, caused either by some FCC families with a cryptic HNPCC mutation, and one or more individuals who had “phenocopy” MSI− cancers with normal expression of mismatch repair proteins,16 or by families or cases with only a single cancer available for analysis which was MSI+ with MLH1 loss, and was therefore indistinguishable from sporadic MSI+ CRCs with biallelic silencing of MLH1 expression.
The similar frequencies of nuclear β-catenin expression in the different CRC series and the absence of an association between nuclear β-catenin and any clinicopathological variable suggest that some form of Wnt pathway dysregulation may be common to all types of CRC, even if not all tumours achieve this in the same way. The cases without nuclear β-catenin localisation may be developing along a different pathway or may represent tumours with more subtle Wnt dysregulation. Previous reports that not all FAP tumours show nuclear β-catenin,24 despite obligate biallelic APC mutation, suggest that the latter is at least partly true.
The fact that the FCC series was consistently divided into two groups by presence or absence of K-ras mutation suggests that this may be a true classifier of familial bowel cancers of unknown genetic origins. Underdiagnosis of HNPCC cannot readily explain this classification, as K-ras mutation and MSI were not associated in the FCC cancers. Data from the multiple adenoma series supported the FCC findings in this regard. There is precedence for a classification of CRCs based partly on K-ras status, given the relatively high K-ras mutation in MAP and the low frequencies in FAP and sporadic MSI+ CRCs, even though the functional basis of these observations is not well understood.
We did not detect the molecular associations previously reported by Abdel Rahman et al.15 In our series, nuclear β-catenin expression was as common in cancers from patients with and without identified mismatch repair mutations; and there was no association between p53 and β-catenin expression in our FCC group. The groups of patients studied by Abdel Rahman et al and us differed somewhat in respect of their molecular classification and numbers, and these factors may help to explain the differences between the studies.
Conclusions
We have found that there is considerable overlap between the genetic pathways followed by colorectal cancers in the known Mendelian syndromes, in cases from a family cancer clinic, and in unselected patients. Specific differences between these pathways are also well established, although they cannot currently be fully explained. We have identified a set of patients with multiple adenomas and no evidence of (attenuated) FAP or MAP, and these patients’ tumours resemble unselected and FCC CRCs more than MAP CRCs. Our data show that a combination of mutation screening, MSI analysis, and immunohistochemistry can identify almost all HNPCC cases; only a small minority of the remaining CRC kindreds from family cancer clinics are true cases of HNPCC. Finally, our results highlight the possibility that K-ras mutation status distinguishes two groups of non-HNPCC patients whose cancers currently have unknown genetic origins.
Acknowledgments
We are grateful to the patients who have taken part in this study and to several histopathology departments for sending tumour specimens.
REFERENCES
Footnotes
-
Competing interests: none declared