Article Text

Abstract
Variations in new splicing regulatory elements are difficult to identify exclusively by sequence inspection and may result in deleterious effects on precursor (pre) mRNA splicing. These mutations can result in either complete skipping of the exon, retention of the intron, or the introduction of a new splice site within an exon or intron. Sometimes mutations that do not disrupt or create a splice site activate pre-existing pseudo splice sites, consistent with the proposal that introns contain splicing inhibitory sequences. These variants can also affect the fine balance of isoforms produced by alternatively spliced exons and in consequence cause disease. Available genomic pathology data reveal that we are still partly ignorant of the basic mechanisms that underlie the pre-mRNA splicing process. The fact that human pathology can provide pointers to new modulatory elements of splicing should be exploited.
- ATM, ataxia telangiectasia
- CERES, composite exonic regulatory elements of splicing
- ESE, exonic splicing enhancers
- ESS, exonic splicing silencers
- hnRNPs, heterogeneous nuclear ribonucleoproteins
- ISE, intronic splicing enhancers
- ISS, intronic splicing silencers
- NAS, nonsense altered splicing
- NF1, neurofibromatosis type 1
- NMD, nonsense mediated decay
- PTC, pretermination stop codon
- SNPs, single nucleotide polymorphisms
- SR, serine arginine
- UsnRNP, uridine-rich small ribonucleoproteins
- gene
- minigene
- mutation
- splicing
Statistics from Altmetric.com
- ATM, ataxia telangiectasia
- CERES, composite exonic regulatory elements of splicing
- ESE, exonic splicing enhancers
- ESS, exonic splicing silencers
- hnRNPs, heterogeneous nuclear ribonucleoproteins
- ISE, intronic splicing enhancers
- ISS, intronic splicing silencers
- NAS, nonsense altered splicing
- NF1, neurofibromatosis type 1
- NMD, nonsense mediated decay
- PTC, pretermination stop codon
- SNPs, single nucleotide polymorphisms
- SR, serine arginine
- UsnRNP, uridine-rich small ribonucleoproteins
The availability of fast sequencing protocols is the basis of an ongoing genetic diagnostics revolution. It is now practical to scan at least the coding sequence of any gene for pathological variation. The effect of sequence variation is generally assumed to depend on its location. In most diagnostic laboratories, sequence variants found after analysis of a gene are classified as pathogenic mutations if they fulfil one of the following criteria:(a) result in the introduction of a stop codon that truncates the protein, for example, frameshifts, and an amino acid alteration to a stop codon, (b) affect an invariant splice consensus sequence, (c) can be demonstrated to be de novo in the patient (that is, not present in parents), or (d) were previously reported as pathogenic in the literature and fulfil any of the above criteria or have supporting data to show that function is impaired. However, a new category may now have to be added. The unexpected complexity of the splicing process, which correctly selects the coding sequences, the exons, from the more abundant non-coding sequences, the introns, has revealed the existence of new splicing regulatory elements difficult to identify exclusively by sequence inspection. Variations in these new elements both in coding and non-coding regions may result in unexpected deleterious effects on precursor (pre) mRNA splicing. This makes distinguishing between benign and disease causing sequence substitutions a challenge for medical geneticists. Recent evidence from various laboratories now indicates that many sequence variations affect pre-mRNA splicing.1–3 These mutations can result in either complete skipping of the exon,4–7 retention of the intron,8,9 or the introduction a new splice site within an exon or intron.10,11 In rare cases, mutations that do not disrupt or create a splice site, activate pre-existing pseudo splice sites consistent with the proposal that introns contain splicing inhibitory sequences.12,13 In addition, these variants can affect the fine balance of isoforms produced by alternatively spliced exons and in consequence cause disease.14 Indeed, possibly the most significant conceptual shift in recent years is that even translationally silent sequence variations should raise suspicion and be seriously considered as being responsible for the pathology observed in the patient.2,15
It is impossible to identify these sequence variations as splicing mutations from genomic data alone unless either they occur in the canonical splice sites or the mutation tracks with disease. With the increasingly widespread use of high throughput sequencing, especially for big genes such as BRCA1, BRCA2, NF1, TSC1, TSC2, and Marfans, many sequence variations of unknown pathological significance will be generated with every plate tested. This can give an ambiguous result for the research scientist, a reporting problem for the diagnostic clinical scientist, and an unsatisfactory counselling problem for the clinician involved. The concern that diagnostically relevant mutations are slipping through the net because their effect on the splicing process has not been considered should now be addressed.
It is not the aim of this review to examine the mechanism of splice site selection or the splicing mechanism, as this subject has being excellently reviewed from different viewpoints over the last few years.16–21 Instead we hope to emphasise the importance of these mutations and the wealth of information they provide along with an overview of the techniques available for assessing their effect on splicing. Available genomic pathology data reveal that we are still partly ignorant of the basic mechanisms that underlie the pre-mRNA splicing process. The fact that human pathology can provide pointers to new modulatory elements of splicing should be exploited. However, a short overview of splicing in general will be necessary before exploring these points further.
PRE-MRNA SPLICING
The general splicing reaction
Before an exon contributes to the coding sequence it has to be included in the mRNA. The removal of introns from pre-mRNA and the joining of exons is a critical aspect of gene expression. This reaction takes place in the spliceosome, which is formed by several snRNP subunits termed uridine-rich small ribonucleoproteins (UsnRNP), and numerous non-snRNP splicing factors. The spliceosome acts through a multitude of RNA-RNA, RNA-protein, and protein-protein interactions to precisely excise each intron and join exons in the correct order.22,23
The splicing reaction takes place in two catalytic steps involving two consecutive trans-esterification reactions24 shown in fig 1. The exact sites for the trans-esterification reactions are defined by consensus sequence, primarily within the intron, around the 5′ and 3′ splice sites.25 The first consensus sequence is called the 5′ splice site and is relatively short. In mammals only the first two bases of the intron (GU) are universally conserved, although they are not the only important bases.26 The second consensus sequence at the 3′ splice site is defined by three separate elements: the branch site, the polypyrimidine tract, and the 3′ splice site dinucleotide AG. Together these elements make up a loosely defined 3′ splice site region, which may extend 100 nucleotides into the intron, upstream to the AG dinucleotide.
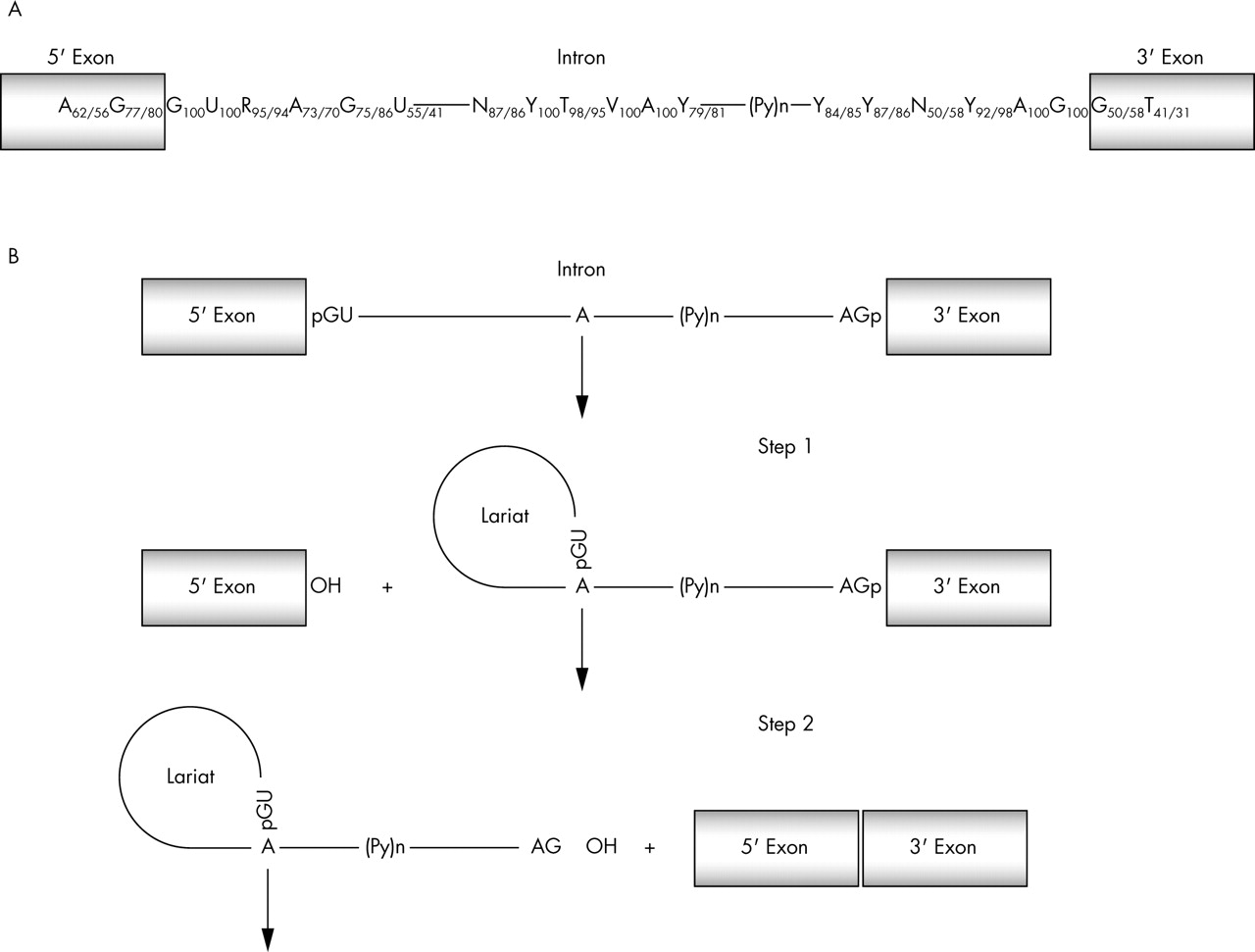
(A) There are several conserved motifs near the nucleotide sequences surrounding the intron-exon borders that act as essential splicing signals. Only the GU and AG dinucleotides, respectively, at the exon-intron and intron-exon junctions along with an A nucleotide in the branch point are universally conserved (frequencies taken from Zhang93). (B) Splicing takes place in two catalytic steps involving two consecutive trans-esterification reactions. During step 1, an adenosine residue generally located within 100 nucleotides of the 3′ end of the intron, in a sequence element known as the branch point sequence, carries out a nucleophilic attack on the 5′ splice site. This reaction generates the splicing intermediates (free exon 1 and lariat-exon 2). During step 2, exon 1 attacks the 3′ splice site to generate splicing products (spliced exon and lariat intron). P(y)n, polyprimidine tract.
Recognition of splice sites
As in its most basic form an exon can be identified by a few cis-acting elements, recognising exons may therefore seem a straightforward linear process. However, the sequences used as 5′ and 3′ splice sites are so diverse that many candidates can be found in a typical mammalian transcript that match these consensus sequences as well as, or better than, the sequences at real splice sites. However, these sequences are not used for splicing. For example, a computer search for such sites in the 42 kb human hprt gene,27 which contains nine exons and eight introns, found the eight real 5′ splice sites but also over 100 5′ pseudo-splice sites that have scores higher than the lowest scoring real internal 5′ splice site. The case was even more extreme for 3′ splice sites, where 683 pseudo-sites were found with higher scores than the lowest scoring real site.
The splicing of most introns thus seems to involve a choice between multiple possible splice sites. In fact, one of the major questions in mammalian pre-mRNA splicing is: how are real splice sites recognised? What is today abundantly clear is that, although necessary, these consensus elements are by no means sufficient to define exon-intron borders.
Enhancers and silencers
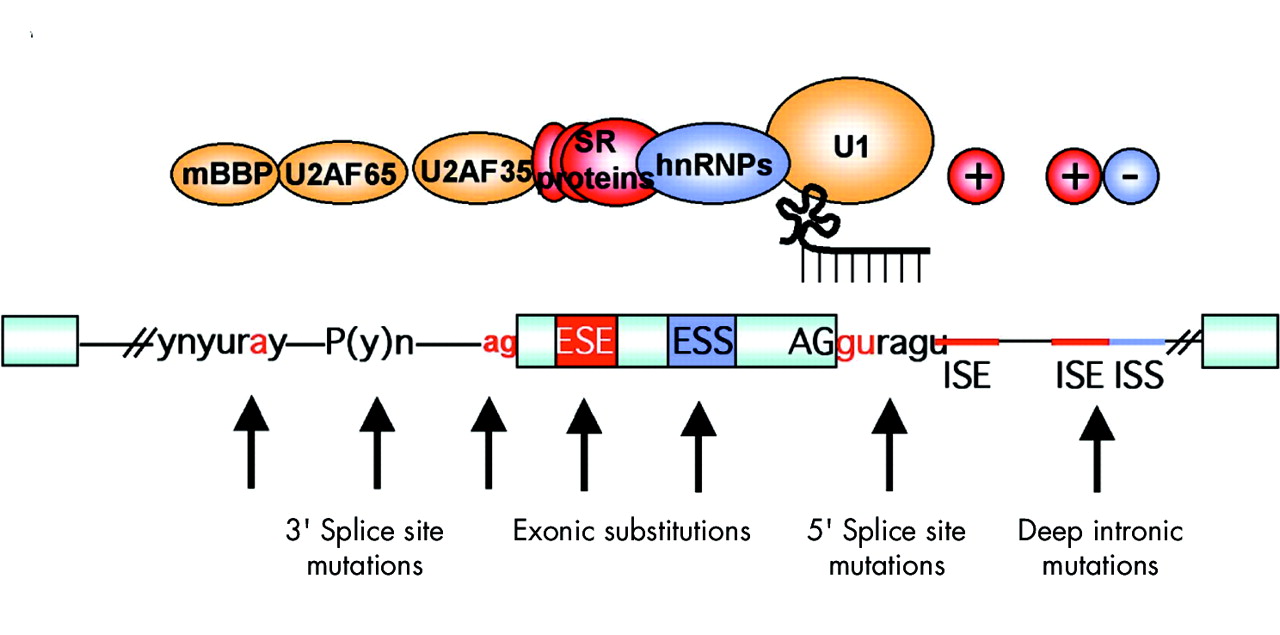
To increase the overall fidelity of the splicing reaction, additional sequences are present in exons and introns. These cis-acting sequence elements can act by increasing or decreasing recognition and are, respectively, named exonic or intronic splicing enhancers (ESE, ISE) or silencers (ESS, ISS). Importantly, any of these sequences could be a target for pathogenetic mutation (fig 2).

Regulatory elements in pre-mRNA splicing and mutations that can affect them. The pale blue boxes represent exons, separated by intervening sequences (introns) shown as lines. Conserved, canonical splice signals (gu, ag) are present at the 5′ and 3′ ends of the exons. These bind the U1 RNA by complementarity and the U2AF35 protein, respectively. If mutations are found in these areas, then the effect on splicing can be assumed to be aberrant. The effect of mutations on the other classical splicing signals upstream of the 3′ splice site, the polypyrimidine tract and the branch point, is less certain, but many examples exist where these can cause incorrect splicing. The trans-acting factors that bind to these regions are U2AF35/65 and mBBP. Additional enhancer and silencer elements in the exons (ESE; ESS) and/or introns (ISE; ISS) allow the correct splice sites to be distinguished from the many cryptic splice sites that have identical signal sequences. Trans-acting splicing factors can interact with enhancers and silencers and can, accordingly, be subdivided into two major groups: members of the serine arginine (SR) family of proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs). In general, but not exclusively, SR protein binding at ESE facilitates exon recognition whereas hnRNPs are inhibitory. Mutations in any of these regions may affect the splicing process due to disruption of the binding of these factors or indeed the creation of a binding site for them.
Enhancers and silencers are involved in both constitutive and alternative splicing and in most cases they lack a well defined consensus sequence. Furthermore, these elements are not always univocally defined and their functions may overlap. In fact, in some systems it may be more appropriate to talk about composite exonic regulatory elements of splicing (CERES) as has been recently described for CFTR exons 9 and 12.28,29
ESE have been the subject of many studies and most, but not all, are known to be recognised by members of the SR protein family.2,30–35 In particular, A/C-rich exon enhancers32 have also been identified through functional SELEX to play an important role in exon definition. These cis-acting elements have been shown to bind and act through trans-acting factors other than SR proteins, for example the YB-1 protein36 and the DEAD-box RNA helicase p72.37 SR proteins bind to ESE through their RNA binding domains and promote splicing by recruiting spliceosomal components through protein-protein interactions by means of their arginine serine rich domains. Further studies have attempted to elucidate the RNA binding specificities of each SR protein.38 Unfortunately, it appears that these proteins recognise a vast array of RNA sequences which are highly degenerate.
The factors that bind to ESS and ISS have not been characterised to a similar extent, however, hnRNPs have been generally implicated in interactions with these elements.39–41 Even less is known about the mechanisms by which ISE work, although in one case GT repeats have been implicated.42
Secondary structure
To complicate matters further, increasing evidence is now available showing that RNA specific secondary structure influences the splicing machinery and plays an important role in exon definition for particular transcripts.43 Although in many of these cases individual peculiarities are involved, the underlying mechanisms by which this occurs can be basically grouped together by the ability of structural elements to hinder or enhance the accessibility of selected sequences by basic splicing factors. This can occur directly, such as in the case of the tau gene44 where the fact that the donor site is present in a stem hinders its use as a splice site, or indirectly, for example by varying the relative distance between these elements.45 Another interesting example involves a specific sequence in the fibronectin EDA exon that has been shown to affect the binding of SR proteins to an ESE which lies 13 nucleotides upstream in the primary RNA. In normal circumstances the function of these sequences has been proposed to stabilise the secondary structure of the ESE in such a way as to allow binding of SR proteins.46,47
MUTATIONS THAT CAUSE ABERRANT SPLICING
Sequence variations occurring in exons or introns may affect the correct processivity of the mRNA by disrupting the splice site, ESE, ESS, ISE, or ISS or altering the secondary structure of the mRNA. Furthermore, in the case of nonsense mutations suspected of affecting splicing, along with disruption of possible ESE and ESS, two other possible mechanisms should also be considered. The first mechanism is known as nonsense mediated decay (NMD) and may explain phenotypic differences among individuals.48 In this case, when a pretermination stop codon (PTC) is present the entire mRNA is degraded.49 As a result of this process the apparent abundance of an exon skipped isoform could be magnified. Nonsense altered splicing (NAS) is more controversial. In this case, a translation-like machinery scans the reading frame and surveys its integrity before splicing. An interrupted reading frame due to a PTC is somehow marked to be skipped, thus maintaining the integrity of the reading frame.50
It is still unclear how frequently sequence variations involving splicing are involved in the onset of disease. In a survey where the mutations considered consisted only of those directly affecting the standard consensus splice sites, 15% of point mutations resulted in a human genetic disease through RNA splicing defects.51 However, in studies of neurofibromatosis type 1 (NF1)52 and ataxia telangiectasia (ATM)53 where analysis was performed both at the DNA and RNA level, approximately 50% of the patients were found to have disease due to mutations that resulted in aberrant splicing. Of these mutations 13% and 11%, respectively, would have been mis-classified only as frameshift, missense, or nonsense mutations if the analysis had been limited to genomic sequence alone and their effect on splicing overlooked. The missense or same sense mutations, in particular, could have been overlooked as being responsible for pathology as the amino acid change may be considered benign. However, as has been shown in the past, these may have dramatic effects on pre-mRNA processing.2,15 Although it seems likely that larger, more complex genes will be more vulnerable to splicing derangements, it is probable that a significant percentage of mutations in many other genes will also be involved in altering the splicing process. Interestingly, most of the splicing mutations identified in the NF1 and ATM studies did not involve the conserved essential splice sites, confirming the suspicion that these mutations would have been overlooked and incorrectly classified. In the case of familial breast and ovarian cancer, approximately 4% of the genetic variants in the BIC (Breast Cancer Information Core) database (http://research.nhgri.nih.gov/bic) are reported as splice site alterations. However, numerous missense sequence variations (accounting for 25% of all mutations reported) have been linked to predisposition to breast cancer, yet the functional significance of the majority of these mutations is unknown, although aberrant splicing has been shown in some.54 In the absence of a functional assay, the biological and pathological significance of many of these missense mutations will continue to remain largely unknown. Thus, in the midst of growing evidence that misclassification of mutations might commonly occur, the general extent of splicing mutations has probably been underestimated. Furthermore, other aspects of transcription could also be affected by changes in gene sequence and are rarely considered. Defects of the initiation, elongation, and termination stages of transcription could in theory be additional potential targets for subtle disease causing mutations.20,55
METHODS TO IDENTIFY SPLICING ABNORMALITIES
Although the volume of genomic information is increasing, the proportion of experimentally confirmed data is not following at the same pace raising the complexity of extraction of useful information. Given the large number of sequence variations found in diagnostic testing where the clinical significance cannot be adequately assessed, it would be useful to be able to study the effect on splicing so conclusions about pathogenicity could be reached. One difficulty of this task stems from our incomplete knowledge of the molecular mechanisms involved. Improvements in this field will invariably help. In a diagnostic setting the methods for assessing splicing would ideally be simple, reproducible, and not involve further sampling from the patient. In fact, studies using RNA extracted from individual patients to confirm splicing defects are not always possible or practical as they can involve potentially difficult RNA extraction from cell lines or tissues, and can be complicated by sources of variability, such as splice site leakiness in some tissues and lower levels of expression of mRNA from mutant alleles. More importantly, in the majority of cases the necessary further samples may be hard to obtain or unavailable to the laboratory.
Alternative techniques, for example using experimental procedures such as functional splicing assays based on DNA along with in silico methods, could potentially circumvent these problems.
In silico tools
Used in conjunction with experimental data, in silico methods provide a useful tool for an initial approach to any mutation suspected of causing aberrant RNA processing.
There are several computer programs available on the web to assess mRNA processing. However, the reliability of such predictions is more difficult to assess and these programs can both over- and under-call the importance of a sequence variation in the splicing process.
Which computer program to use obviously depends on the location of the sequence variation. One of the few exceptions for which no in silico predictions are available is the case of deep intronic mutations and in this case the only possible way to see if these affect splicing is to assess them experimentally.
Splice site mutations
Most disease causing single nucleotide substitutions in donor or acceptor splice sites involve the +1/+2 or −1/−2 position, respectively.51 However, pathological splicing alterations have also been observed to involve a variety of other positions, which can be close or fairly distant from these two pairs of canonical nucleotides. The functional reason why alterations in positions close to the splice sites may cause aberrant splicing is usually due to the disruption of interactions with U1 snRNP, U6 snRNP, and U2AF 65 or 35 in the splicing process.56
Instances of point mutation within the branch point sequence have also been published, and do not necessarily occur at the invariant adenosine in the consensus branch site sequence which is required for lariat formation.57
The difficulty when considering sequence variations surrounding the canonical splice sites for their effect on splicing lies in the fact that these intronic cis elements (5′ splice site, 3′ splice site, and branch site) are weakly conserved. This degenerative nature means that alterations in these nucleotide positions do not always disrupt the splicing processes. There are several web sites that analyse the strengths of splice sites and the effect of different sequence variations on them. The more widely used are:
Although all are valid, the first program in this list uses 15 (−7 to + 8) bases to calculate a score for a 5′ splice site and 41 bases (−21 to +20) to calculate a score for a 3′ splice site. The large number of nucleotides utilised means that variations quite distant from the canonical dinucleotides can be assessed.
These programs cannot distinguish between pseudo and real splice sites. This means that if a mutation is being analysed for its ability to create a cryptic site, a functional test must be performed.
ESE mutations
Nonsense, missense, and silent mutation induced exon skipping is recognised in an ever increasing number of genes as a novel form of splicing mutation. Evidence exists1–3,14,58 that the disruption of ESE sequences is frequently the mechanism underlying these types of mutation associated exon skipping. Two ESE prediction programs are currently available: ESEfinder at http://rulai.cshl.edu/tools/ESE/ and RESCUE-ESE at http://genes.mit.edu/burgelab/rescue-ese.
The first of these, ESEfinder, a web based tool now out in its second edition,59 provides sequence weight matrices for scoring a subset of candidate ESE motifs corresponding to the functional consensus motifs of four SR proteins that were identified through a functional SELEX method.38 The predictive value of these matrices is exemplified by the fact that high score motifs are enriched in exons, clustered in regions containing natural ESE,2,4,60 and by correlations between motif scores and exon skipping phenotypes in several genes.61 A possible drawback of this program is that it searches the ESE motifs corresponding to only four SR proteins. This means that sequences corresponding to the RNA binding specificities of other SR proteins (of which there are at least 10) or for other types of proteins, will be missed by the program.
RESCUE-ESE predicts motifs with ESE sequences based on the statistical analysis of differences in hexamer frequencies between exons and introns and between exons with weak and strong splice sites.62 The web server allows a sequence to be checked for the presence of these candidate ESE hexamers. Though less corroborated than ESEfinder, this program has also resulted in the correct identification of sequences that act as ESE and representatives of 10 predicted motifs have been shown to display enhancer activity in reporter minigenes.63
Both programs have been tested by their creators and by external users and there are many prediction success stories. However, the ability of both programs to predict natural point mutations that disrupt genuine ESE motifs has yet to be fully assessed. A real count of false positives, that could be quite high as in the majority of cases they go unpublished, is still lacking. Another point to keep in mind is that exon definition is an extremely intricate process. For this reason, the presence of a high score motif or indeed an ESE motif in a sequence does not necessarily identify that sequence as an exonic splicing enhancer in its native context. For example, nearby silencer elements may prevent the sequence acting as an enhancer or the sequence may be part of an element such as the recently described CERES28,29 where overlapping enhancer and silencer functions exist which are not completely dependent on SR protein interactions.
One possible way of improving the predictive power of these tools when applied to a particular exonic sequence is to additionally assess the strength of the splice sites of the exon. If either the 5′ or 3′ splice site proves to be weak, then the probability of the in silico prediction of an ESE being correct would increase. However, it is very important to stress that until more refined prediction algorithms have been developed, experimental data either from patient samples or minigene systems remain indispensable to assess the clinical significance of the genomic variation.
Secondary structure prediction
Changes in RNA structure have been invoked as playing a role in pathogenetic processes involving the dystrophin gene,64 the NF1 gene,65 and, more recently, the CFTR gene.66 In humans, secondary structures which affect splice site consensus sequences have also been described in the generation of human growth hormone isoforms,67 tau,44,68–70 HPRT,71 and the hnRNPA172 genes. Recently, the fact that most major members of the SR protein family have been observed to be potentially affected by the conformation of a target RNA may indicate that structural influences may be a widespread occurrence at least for the components of this important family of splicing modifiers.46
The alteration of pre-mRNA secondary structure is potentially applicable to all kinds of single nucleotide substitutions that lead to exon skipping as single nucleotide polymorphisms (SNPs) are capable of inducing in vivo different structural folds in mRNA structures.73 However, the effect of these SNPs on splicing or function has yet to be tested.
Various secondary structure prediction programs exist on the web. Those principally used are Mfold, based on free energy minimisation,74,75 and Pfold, based on an evolutionary probabilistic model.76,77 The drawback of these approaches is the fact that computer algorithms provide a folding prediction (often more than one) for virtually any RNA sequence and are strongly biased by the length of the RNA sequence examined. For this reason, although in silico predictions are an invaluable tool for the researcher in this field, special care should be exercised when predicted pre-mRNA structures are correlated with splicing behaviour. As an example, see the recent application to the NF1 gene,65 implicated in the generation of human tumours, where the predictive power of stand alone in silico approaches has been challenged.78 Evidence from experimental structural probing that the proposed structure follows structural predictions should always be checked. The fact that this is often not the case emphasises the fact that this experimental procedure is rather complicated, and as such is not outlined in this review. However, a comprehensive overview of this methodology can be found in Buratti et al46 and Muro et al.47
FUNCTIONAL SPLICING ASSAYS
The definitive test of whether a suspected disease causing mutation affects splicing ideally comes from RNA analysis of affected tissue, as cis-acting splicing mutations can have cell specific effects. Unfortunately, the appropriate tissues are often not available. As an alternative, the mutation can be definitively identified using transient transfection of minigenes or in vitro splicing assays, comparing the splicing patterns of mutant and wildtype exons. These types of assays can also be used to further explore the mechanism behind aberrant RNA processing.
A minigene, as its name indicates, is a simplified version of a gene. Its first use dates back about 20 years to experiments providing the first structural and functional evidence that the primary transcript of fibronectin undergoes alternative splicing,79 and that exonic sequences are involved in splicing modulation.80 Modified versions of the same construct are still used today in unravelling novel mechanisms of splicing when used to test if mutations observed in pathological cases are splicing mutations. Uses also include the identification of the CERES element,28,29 deep intronic splicing modulators,13 and the role played by donor site mutations.6,81
The minigene used in these cases is a hybrid construct containing exons from α-globin and fibronectin, under the control of the α-globin promoter (fig 3A). The intronic region between the two fibronectin exons contains a unique NdeI site. In the case of exonic mutations suspected of affecting splicing or of affecting intronic mutations near the exons, the exon along with a small amount of flanking intronic sequences (∼200 bp) can be inserted in the NdeI site. Deep intronic mutations can be analysed with this system by creating hybrid minigenes in which the two exons flanking the intron carrying the mutation and the intron itself are inserted into the minigene at the NdeI site (fig 3B).

(A) Schematic representation of minigene splicing assays. Mutations suspected of being involved in aberrant splicing can be analysed using a hybrid minigene. A typical hybrid minigene is a plasmid that contains a “simplified” version of the gene that will be evaluated for pre-mRNA splicing. This example contains at the 5′ end an α-globin gene promoter and SV40 enhancer sequences (indicated by the arrow at the start of the gene) to allow polymerase II transcription in the transfected cell lines. This is followed by a series of exonic and intronic sequences (indicated by boxes and lines, respectively) that may derive from a reporter gene or from the gene context itself. In this case, the reporter gene is composed of α-globin (black boxes) and fibronectin exons (grey boxes), while at the 3′ end a functionally competent polyadenylation site, derived from the α-globin gene, is present. The genomic DNA region of interest that contains a putative splicing mutation is introduced into the minigene in a unique restriction site (NdeI). (B) In the case of deep intronic mutations, hybrid minigenes are created in which the two exons flanking the intron carrying the mutation and the intron itself are inserted into the minigene at the NdeI site. Pale grey circles indicate regions of possible mutations that need to be analysed. Black horizontal arrows represent primers used for PCR amplification. (C) Schematic representation of the hybrid minigene (SXN13). This minigene consists of a 34 nucleotide alternative exon flanked by duplicated intron 1 from human α-globin such that the first and third exons are globin exons 1 and 2. In the absence of a splicing enhancer this element is predominately skipped due to its small size and a non-canonical 5′ splice site. Regions of exonic DNA suspected of having enhancer activity can be cloned into the alternative exon and tested for their effect on splicing.
Following transfection into cultured cells, where the minigenes are transcribed and spliced, the mRNA derived from the hybrid minigene is analysed using reverse transcriptase PCR (RT-PCR) with oligonucleotides that amplify only the exogenous product as they are complementary to the sequences overlying the junctions of the hybrid exons and thus do not exist naturally. Finally, the spliced products are visualised on an agarose gel (fig 4).
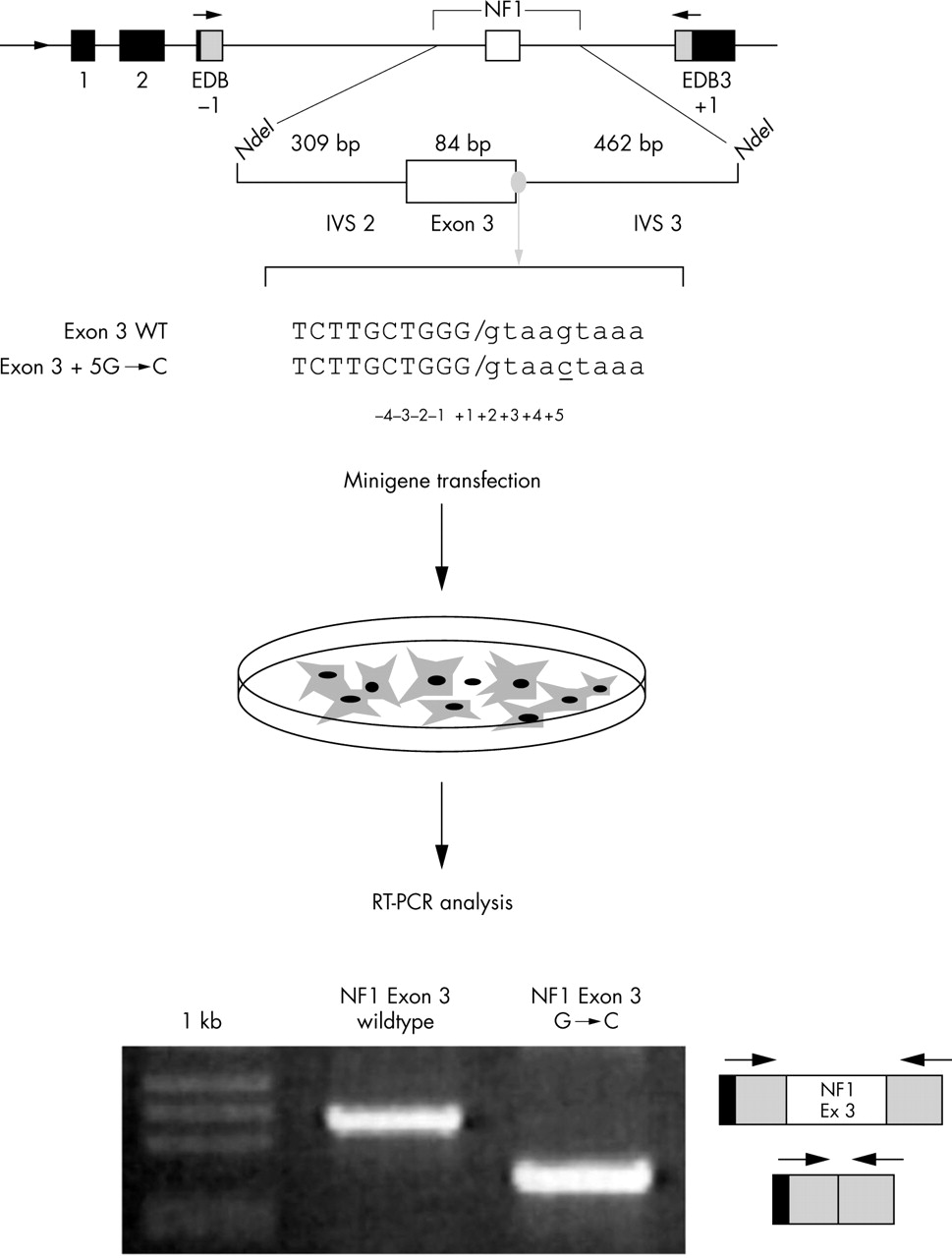
Functional splicing assay. In the upper part of the figure is a schematic representation of the α-globin fibronectin NF1 exon 3 minigene created in order to test a nucleotide variation in the +5 position of exon 3 for its effect on the splicing process.6 Exonic sequences are in upper case and intronic sequence in lower case. The mutation is underlined. The minigenes (wildtype and mutated) are transfected in appropriate cell lines, followed by RNA extraction, RT-PCR, and gel electrophoresis. In this example, analysis of the transcript derived from the wildtype minigene (exon 3 WT) shows the correct inclusion of an exon, whereas the transcript derived from the mutated minigene (exon 3+5 G→C) does not include exon 3 as indicated schematically on the right of the gel.
The principal disadvantage of this minigene is that in an ideal system the surrounding introns and exons would come from the same gene. However, this is also the principal advantage of the system. In fact, in order to test the effect of a mutation on splicing, a minimum of a three exon, two intron system is necessary and in most genes this would involve handling several thousand nucleotides. However, with this type of minigene only short DNA fragments need to be handled. Another factor that needs to be considered is that when analysing the effect of NAS or NMD it is important that the correct reading frame is utilised.
Wherever possible three exon, two intron systems composed from the gene under study should be utilised. These can also simply be made by cloning the DNA fragment into a commercial vector such as pcDNA 3. This vector carries a CMV promoter and the appropriate stop signals of transcription. The DNA fragment can be put in phase, if this is not already the case, by the addition or subtraction of the appropriate number of nucleotides as well as the addition of a Met initiation codon for the start of translation through PCR mutagenesis.
A variation of these minigenes, and possibly of greater interest for high throughput testing of exonic mutations thought to disrupt ESE, is exemplified by that utilised in the identification of ESE by in vivo selection.32,63 This hybrid minigene (SXN13) consists of a 34 nucleotide alternative exon flanked by duplicated intron 1 from human α-globin such that the first and third exons are globin exons 1 and 2 (fig 3C). The translation start codon has also been deleted in order to avoid exon skipping occurring through inadvertent introduction of a nonsense codon that could result in aberrant splicing through NMD or nonsense associated altered splicing (NAS). In the absence of a splicing enhancer this element is predominately skipped32 due to its small size and a non-canonical 5′ splice site, however low levels of enhancer independent inclusion do occur. This exon contains a small cassette into which 13 nucleotides of choice can be cloned, and the enhancer ability of the sequence tested. The reasoning behind the system is that sequences with ESE capacity would have the ability to boost inclusion of this weakly recognised exon. This system however suffers from the fact that efficient splicing is a result of a number of complex and often antagonistic and/or synergistic interactions mediated by different splicing elements that can occur across both introns and exons.33,82,83 Therefore, although a system such as this could indeed give an indication that the mutation under study is disrupting an ESE, whether the mutation alone is responsible for the pathology would need further investigation. However, systems based on this type of minigene developed for mass testing of suspected splicing mutations could be very useful.
An alternative to the methodology described above are the in vitro splicing assays. Here, labelled pre-mRNA transcribed with bacterial polymerases is incubated in the presence of nuclear extracts and the resulting spliced products resolved on a polyacrylamide denaturing gel. The real advantage of this system is that splicing intermediates can be evaluated and molecular mechanistic studies could be performed by depleting the nuclear extract of various proteins. For an application of this technique, see Buratti et al.84 However, with the advent of SiRNA technology this last technique can now be readily applied to cells in culture. The main disadvantages are that the system allows only relatively short sequences to be studied and does not take into account the recent observations that in some instances transcription and splicing are intimately connected in the cell.20,85,86
PRACTICAL EXAMPLES OF IN SILICO AND EXPERIMENTAL APPROACHES TO IDENTIFY SPLICING MUTATIONS
Table 1 lists 67 NF1 mutations found by molecular diagnostic testing in patients diagnosed with NF1. The table is broken down into the classical mutation categories and a database and literature search was carried out in order to identify previously know mutations which had been found to affect pre-mRNA splicing. Out of the 67 mutations identified, only one had been previously shown to affect splicing (Y226X). If we add this to the mutations/deletions occurring in the canonical splice (AG-GT) nucleotides, which are certain to result in incorrect splicing of the IVS in which they are found, the percentage of splicing mutations present is approximately 13%. This number is well below the 50% suggested as the number of pathological mutations in NF1 to affect splicing.52
Mutation analysis results on the NF1 gene with in silico analysis of the sequence variations found for splice site strength and presence of ESE
In silico tools for splice site strength and the presence of ESE were then utilised to identify other mutations affecting splicing.
Table 1 lists a series of possible splice mutations where the nucleotide substitution occurs in the area surrounding donor and acceptor splice site consensus sequences. The use of the splice site strength prediction to some extent provides us with further clues as to which mutations may affect splicing, as in some cases the substitution causes the splice site strength score to fall drastically. If we consider these as splice site mutations and add them to those in the AG-GT nucleotides, approximately 20% of the mutations can now be classified as splicing mutations. However according to the statistics previously calculated,52 up to a further 20 mutations in this list could still be categorised as splicing mutations.
For convenience we decided that all the mutations in the splice site consensus that lowered the score below the threshold resulted in aberrant splicing. However, some of the mutations in the splice site consensus did not drastically affect the splice site strength score. For example, the vicinity of the nucleotide change in exon 3 to the 5′ splice site of intervening sequence 3 suggests that it may interfere with the splicing of exon 3. However, the 5′ splice site strength score of the mutant allele is much higher than for many wildtype donor scores. Indeed, examples of wildtype sequences similar to the exon 3+5 G→C mutation in which the corresponding exon is spliced efficiently can be easily found: for example, within the normal NF1 gene, IVS 1 and 7 share an identical –1 to +5 sequence with the mutated IVS 3, yet splice normally. This case is a good example of the problems encountered in mutation classification when using simple sequence analysis alone. We did however classify this mutation as disease causing as it was observed in the proband and her granddaughter but not in 100 normal chromosomes.87 Subsequent minigene expression studies and addition of a modified U1 snRNA complementary to the observed mutation allowed us to categorically classify this as a splicing mutation which acted by causing the abrogation of binding of U1 snRNP to the 5′ splice site resulting in exon 3 skipping.6,81
Same sense, missense, and nonsense mutations may, as discussed above, be responsible for aberrant splicing due to abrogation of ESE and ESS.1–3,14,58 From table 1 we can see that these categories of mutation are the majority of those found in diagnostic screening of NF1. As it is probable that some of these also interfere with splicing, we investigated which of these could possibly disrupt ESE sites by analysing them with the two available ESE prediction programs. These predictions are summarised in table 1 where the creation or disruption of an individual SR protein site (calculated by ESEfinder) or hexamer sequence (ESE-RESCUE) by the mutation is reported. Unfortunately, the data that emerge from the application of these in silico tools is confusing. Either or both programs predicted the disruption of ESE in 18 out of 35 mutations, although 31 out of the 35 mutations were predicted to affect an ESE (creation or disruption). Moreover in several instances both ESEfinder and ESE-RESCUE predicted the disruption of one ESE site concomitantly with the creation of another. Furthermore it is important to note that the table does not report increases or decreases in ESE scores as calculated from ESEfinder. These results confirm previous observations indicating that ESE prediction programs tend to overestimate the number of mutations that may cause exon skipping. However they also show that any data inferred from these algorithms need to be tested experimentally, the ideal systems being a minigene.
The mutation Y226X in exon 37 is one such example of the problems that can arise with in silico data analysis. Admittedly the stop codon created immediately classifies this mutation as disease causing. However, the real mechanism by which the pathology occurs is not through the production of a truncated protein at this exon but through skipping of the exon. In fact previous studies have shown that this mutation causes in frame skipping of the exon, raising the possibility of a NAS mechanism being active.88 However, other mutations89 found within a few base pairs of Y226X and causing stop codons, exert quite different effects on splicing with the processing of mRNA remaining normal (fig 5). These results indicate that exon skipping is apparently due to the mutation itself possibly disrupting an ESE. The results of the two ESE programs found a reduction in a high motif for SRp40 (4.8 to 2.8) and creation of a SRp55 motif according to ESEfinder, and creation of a novel ESE hexamer according to ESE-RESCUE. A further interesting observation is that this exon has a sub-optimal 3′ splice site, possibly indicating the need for an ESE in the exon to allow the splicing machinery to recognise it as such.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Localisation of different stop mutations within exon 37 of the NF1 gene and their effects on pre-mRNA processing. The figure shows a short section of NF1 exon 37. The stop codon caused by the substitution of the nucleotide at position 6792 for either a G or an A creates stop codon TAG or TAA, respectively, that leads to skipping of exon 37. In contrast, mutations 6790ins TT and 6789del TTAC cause a frameshift that, in both cases, results in a stop codon at position 6806 6808 (TGA). However, a stop codon at these positions does not interfere with the subsequent splicing process.89(B) Localisation of the same sense (translationally silent) mutation in position 6792 of the NF1 gene created by substituting the C for a T. This nucleotide change was observed by us to cause exon skipping (unpublished data).
Through the use of a minigene constructed by cloning NF1 exons 34–38 into pcDNA3, we observed that even a same sense (translationally silent) mutation in this position (6793) caused exon skipping (fig 5B), indicating that aberrant processing of mRNA carrying this mutation would indeed be due to the disruption of an ESE rather than NAS. However, through immunoprecipitation with antibodies against the majority of SR proteins, it would appear that this ESE does not interact directly with SR proteins, pointing towards a mechanism different from that suggested by ESEfinder (our unpublished data).
CONCLUSIONS
With the increasing recognition that sequence variations frequently affect splicing by using mechanisms other than just disrupting splice sites, this form of gene mutation must now be considered in greater detail. The data associated with the effect of silent sequence variations and splicing are particularly impressive.4,90
It is no longer possible to ignore these types of sequence variations, however innocent they may look, until a possible effect on splicing has been considered. Similarly, missense sequence changes are commonly found when analysing large genes and can also create diagnostic dilemmas. Furthermore, these cases will be very useful in clarifying the basic molecular mechanisms underlying the pre-mRNA splicing process if studied in more detail.
In the diagnostic environment, assays for assessing splicing would be a useful adjunct to the service. In particular, we have shown that in silico assays, although useful in giving a very preliminary indication of the effect of a sequence variation, currently need to be complemented by further analysis, for example by using a minigene splicing assay. Even the authors of the programs (such as ESEfinder and RESCUE-ESE) strongly suggest this line be followed. In the midst of excitement, however, this recommendation can fall on deaf ears, with a number of reports being published that state that the program has been run and conclude that their sequence variation of choice is affecting splicing purely because an in silico program said so, followed by subsequent publications that carry experimental evidence contradicting these theories.91,92 Indeed, in some cases the actual mechanisms underlying the aberrant splicing may be extremely complex and involve a series of regulatory factors such as appears to be the case for the SMN gene.4,90
Minigene testing itself need not be complicated and could be adapted for use in a diagnostic molecular laboratory, using DNA samples already available. Our increase in knowledge of the basic mechanisms of splicing provided by functional experiments such as these are an essential step to an improvement of in silico assays.
Finally, it has been suggested that the mutations currently described as affecting exonic or intronic regulatory elements and causing severe splicing defects, might just be the tip of the iceberg. Some sequence variations could cause partial splicing defects that are only pathogenic in specific tissues under the influence of a set of specific regulatory splicing factors and/or specific genetic background.
Acknowledgments
We thank Professor Baralle for his helpful comments and reading of the manuscript.
REFERENCES
Footnotes
-
Competing interests: none declared
-
The numbers used to designate the mutations refer to the coding sequence of the NF1 gene (accession number NM 000267). The symbols + or – designate upstream or downstream, respectively.