Article Text

Abstract
Background De novo mutations in PURA have recently been described to cause PURA syndrome, a neurodevelopmental disorder characterised by severe intellectual disability (ID), epilepsy, feeding difficulties and neonatal hypotonia.
Objectives To delineate the clinical spectrum of PURA syndrome and study genotype-phenotype correlations.
Methods Diagnostic or research-based exome or Sanger sequencing was performed in individuals with ID. We systematically collected clinical and mutation data on newly ascertained PURA syndrome individuals, evaluated data of previously reported individuals and performed a computational analysis of photographs. We classified mutations based on predicted effect using 3D in silico models of crystal structures of Drosophila-derived Pur-alpha homologues. Finally, we explored genotype-phenotype correlations by analysis of both recurrent mutations as well as mutation classes.
Results We report mutations in PURA (purine-rich element binding protein A) in 32 individuals, the largest cohort described so far. Evaluation of clinical data, including 22 previously published cases, revealed that all have moderate to severe ID and neonatal-onset symptoms, including hypotonia (96%), respiratory problems (57%), feeding difficulties (77%), exaggerated startle response (44%), hypersomnolence (66%) and hypothermia (35%). Epilepsy (54%) and gastrointestinal (69%), ophthalmological (51%) and endocrine problems (42%) were observed frequently. Computational analysis of facial photographs showed subtle facial dysmorphism. No strong genotype-phenotype correlation was identified by subgrouping mutations into functional classes.
Conclusion We delineate the clinical spectrum of PURA syndrome with the identification of 32 additional individuals. The identification of one individual through targeted Sanger sequencing points towards the clinical recognisability of the syndrome. Genotype-phenotype analysis showed no significant correlation between mutation classes and disease severity.
- PURA syndrome
- intellectual disability
- neonatal problems
- hypotonia
- epilepsy and seizures
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Deletions in the 5q31.2q31.3 region have been reported to cause a syndrome comprising severe developmental delay, profound hypotonia, feeding difficulties, abnormal breathing pattern and seizures.1–5 Within this region, haploinsufficiency of the purine-rich element binding protein A (PURA) has been suggested as responsible for the observed neurodevelopmental features by narrowing the region of overlap to three genes.4 Further evidence for the essential role of PURA in development came from two articles describing 15 individuals with intellectual disability (ID) and de novo mutations in PURA.6 7 The pathogenicity of mutations in PURA was further supported by publication of a cohort of six additional individuals with ID and de novo mutations in PURA and two case reports.8–10
The functionality of PURA is dependent on three PUR motifs, PUR I, PUR II and PUR III.11 12 The ubiquitously expressed PURA protein, Pur-alpha, has multiple regulatory functions in processes such as DNA replication and transcription, mRNA transport and DNA repair.12 13 Several studies have shown that Pur-alpha has an important role in neuronal development and differentiation.14 In animal models, Pur-alpha is involved in postnatal brain development.15 16 The Pur-alpha deficient knockout mouse model has a severe neurological phenotype, including tremor and spontaneous seizures.16 Recently, heterozygous PURA knockout mice have been reported to develop gait abnormalities, hypotonia and memory deficits. Immunohistochemical assays displayed a reduced number of neurons in cerebellum and hippocampus of these animals, which is in line with their neurological, behavioural and cognitive phenotypes.17
Mental retardation, autosomal dominant type 31[MIM616158], or ‘PURA syndrome’, the name of the syndrome adopted by the parents and clinical community alike, is phenotypically hallmarked by a wide spectrum of neurodevelopmental problems, including severe neurodevelopmental delay, epilepsy, abnormal movements, hypotonia and brain abnormalities. Additionally, neonatal respiratory insufficiency and feeding difficulties have been reported. Outside of this core phenotype, a broad variability in clinical severity has been observed within PURA syndrome.6–9 Different types of mutations have been identified so far and mutations occur throughout the gene. It has been suggested that disruption of PUR repeat III possibly results in a more severe phenotype, but additional studies are needed to better understand the genotype-phenotype correlation of PURA mutations.7
We present 32 individuals with de novo mutations in PURA. With this large cohort, we delineate the clinical spectrum of PURA syndrome. Computational modelling shows that subtle overlapping facial dysmorphism is present. Additionally, we present an approach for genotype-phenotype analysis of identified mutations and associated phenotypes, revealing that observed phenotypic variability is probably not associated with type and localisation of mutations.
Methods
Identification of PURA syndrome individuals and collection of clinical and mutational data
Whole exome sequencing (WES) was performed in individuals with ID in either diagnostic (n=20) or research (n=10) settings (online supplementary methods). In one individual, targeted massively parallel sequencing (MPS) of a severe childhood epilepsy gene panel was used, and in a second individual, clinically suspected to have PURA syndrome, PURA [NM_005859] was investigated with targeted Sanger sequencing. Parental samples were tested to assess de novo state. A questionnaire with clinical and mutational details was completed by clinicians. Written consent for publication of photographs was obtained from legal guardians.
Supplementary methods
Evaluation of previously published cases
Twenty-two individuals with PURA mutations have been described in four different publications.6–9 We used the same questionnaire to collect clinical and mutation data from individuals (n=22) reported in the literature.
Computational analysis of facial photographs
To visualise the characteristic facial features of PURA syndrome individuals, we generated a realistic, deidentified average based on 34 photographs of individuals (in this paper presented and previously published photographs) at different ages. A control average was created from 301 age-matched, healthy individuals. We used a fully automated algorithm that (1) annotated a constellation of 68 facial landmark points on the face, (2) created an average face mesh and (3) warped faces of each individual onto the average face mesh. The face averaging algorithm was inspired by previous work18 19 and improved upon with better deidentification of individuals (online supplementary methods). Interpretation of the composite PURA face was performed by a panel of five independent dysmorphologists.
Functional assessment of PURA mutations in structural homology models
For homology modelling of mutations, the crystal structures of the N-terminal PUR domain (PDB-ID: 3K44) and of the C-terminal PUR domain (PDB-ID: 5FGO) from Drosophila melanogaster were used as template. Homology structures of human PUR domains were calculated using the PHYRE2 program.20 In the case of the C-terminal PUR domain, the sequences of two repeat IIIs were merged into one peptide chain to allow for modelling of the entire domain. The human homology models showed high confidence scores and were used for in silico mutational analyses using the program Coot.21 All mutations identified in our cohort and previously published cohorts6–9 were included. Variants in PURA reported in the Exome Aggregation Consortium (ExAC) database22 were included as controls. Deletions, frameshift, non-sense and missense mutations were manually classified depending on the position, size and function of the affected amino acids. Point mutations were in silico introduced using the standard rotamer library of Coot and analysed for the appearance of steric clashes or repulsive forces of side chains. They were classified based on the orientation of mutated side chains and on our knowledge of functionally important surface regions. Figures were prepared using the program Pymol (www.pymol.org).
Results
Identification of 32 individuals
We report 32 individuals with mutations in PURA (figure 1). All, but two, were identified using WES in a diagnostic setting (individuals 1–10, 20–27, 30 and 32) or research setting (individuals 12–19, 29 and 31). Individual 28 was identified using targeted MPS of a severe childhood epilepsy gene panel. Individual 11 was clinically suspected to have PURA syndrome based on post-term birth, severe neonatal hypotonia, hypersomnolence, exaggerated startle, persistent apnoeic episodes requiring continuous SpO2 monitoring and supplementary oxygen, and feeding difficulties warranting nasogastric tube placement. Targeted Sanger sequencing revealed a PURA mutation. Mutations were de novo in 29 individuals. For the remaining three individuals, no parental blood was available for testing. None of the individuals had a second, likely pathogenic, genetic variant in WES data. All identified missense mutations localised in one of the PUR repeat sequences (figure 1).
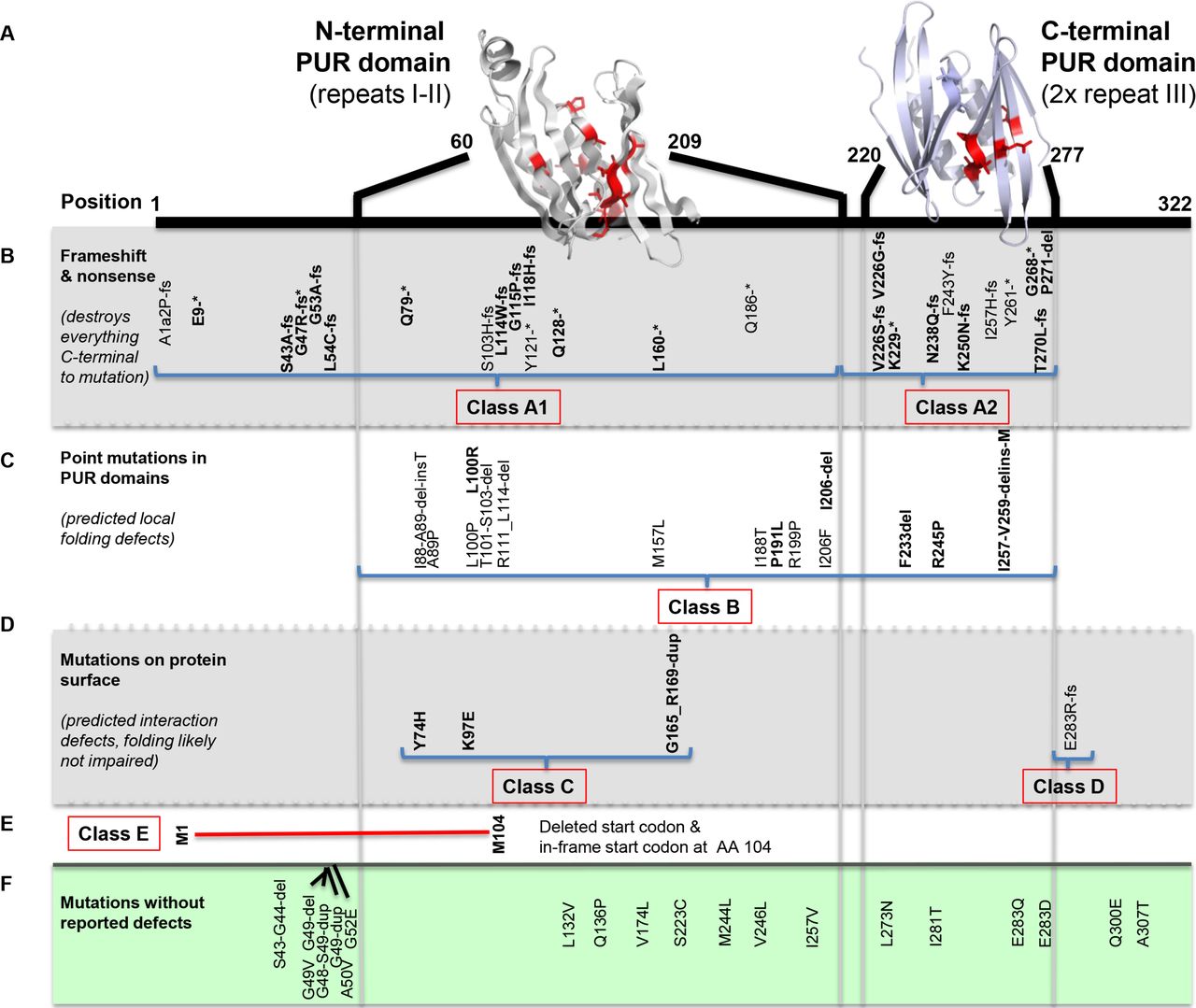
Localisation of PURA mutations and subdivision in classes. Mutations of individuals identified in our cohort are marked in bold. (A) Homology models of N-terminal and C-terminal PUR domains (grey and blue, respectively) from human Pur-alpha. Residues with single amino acid exchanges are depicted in red with side chains. (B) Location of reported PURA frameshift and non-sense mutations. Class A1 mutations are located in the N-terminal PUR domain that affect both N-terminal and C-terminal domains and class A2 mutations occur in the C-terminal domain, affecting only this domain and the C-terminus. (C) Identified point mutations in one of the PUR domains, predicted to cause local folding defects. (D) Four mutations of amino acids located on the protein surface. Three mutations are predicted to affect nucleic acid binding (class C) and one mutation (class D) likely affects a surface-exposed residue, which is not predicted to impair protein folding or nucleic acid binding. This mutation is possibly involved in not yet understood functions such as protein–protein interactions. (E) Deletion (red) caused by mutation p.(Met1?) (class E), which disrupts the start codon. The protein is likely expressed from the next in-frame start codon at amino acid 104, causing loss of a functional N-terminal PUR domain, but has an intact C-terminal PUR domain. (F) Localisation of mutations reported in healthy controls in the ExAC database. All these mutations are predicted to have no effects on the folding and the function of the Pur-alpha protein. PUR, purine-rich element.
Clinical delineation of 32 individuals
Clinical features observed in individuals reported in this study are summarised in table 1. Numbers and percentages of features were corrected for the number of individuals without available information on a specific feature. More extensive and detailed clinical information is available in online supplementary table 1.
Supplementary table 1
Percentage of clinical features reported in individuals in this article (n=32) and meta-analysis with previously published PURA individuals (n=22)6–9
Gestation and neonatal problems
More than half (56%) of individuals were born after >41 weeks’ gestation. Induction of labour and/or caesarian section were reported in 14 individuals. In 55% (6/11), excessive hiccups were mentioned in utero. Neonatal problems were evident in all children immediately after birth. Hypotonia was present in all but one of the neonates, often causing feeding difficulties (81%). Apnoeas and congenital hypoventilation were reported in 48% of the neonates. Both feeding and respiratory problems often required monitoring in hospital: in 20 neonates, tube feeding, oxygen supplementation and/or mechanical ventilation were needed. Hypersomnolence (66%) and hypothermia (37%) were observed in a significant number of neonates. In about half of the neonates (58%) an exaggerated startle response was noted.
Development
All individuals with PURA mutations have moderate to severe ID with severe language and motor delay. Most individuals remain non-verbal (29/32, 91%), but many have better receptive language than expressive language and can follow simple instructions. Severalnever achieved independent ambulation (10/24 with age >5 years, 42%). For those who did, the age of first steps ranged from 28 months to 7 years. Regression of achieved skills due to the onset of seizures has been reported in six individuals.
Neurological abnormalities
A variety of neurological problems were observed in individuals with PURA mutations. Hypotonia, often more prominent in the trunk, was present from birth. Spasticity of extremities at older age was reported in three individuals, and Babinski response was reported in another two. Gait, if achieved, was often unstable and broad based. Stereotypic hand movements, in some cases such as described for Rett syndrome [MIM312750], were observed in several individuals (36%). Six out of 30 (20%) were mentioned to have a movement disorder, including dystonia, chorea-like movements, seizure-like movements with normal EEG and ataxic movements. Delayed myelination is the most frequently reported brain abnormality observed on MRI images (28%). Other, mostly a-specific, observed brain abnormalities (in 31% of individuals) include white matter abnormalities, prominent periventricular spaces, mild parenchymal atrophy, widening of lateral ventricles and underdeveloped rostrum of the corpus callosum. Peripheral neuropathy was measured in five individuals at young age (online supplementary table 1).
Half of the individuals (50%) were diagnosed with epilepsy. For most of them, seizures started at the age of 2–4 years, but the age of onset ranged from 6 months to 15 years. Different seizure types were reported, including generalised tonic-clonic seizures, focal seizures, absence seizures, epileptic spasms, tonic seizures, drop attacks and over time, evolution to Lennox-Gastaut syndrome. The epilepsy is often refractory to medical treatment and some individuals have never been seizure free. A longer follow-up period will be required to determine the true prevalence of epilepsy, how refractory it is and what treatments are most effective.
Ophthalmological abnormalities
Eye abnormalities and visual problems were described frequently in PURA syndrome individuals (40%). Strabismus (often oesotropia) was the most commonly reported finding along with strabismus-associated refractive errors (often hypermetropia). Cortical visual impairment was reported in three individuals; however, it is important to note that diagnostic criteria for this condition are highly variable. One individual was treated successfully for a congenital retinoblastoma. Nystagmus was also described in a small number of cases (4/29; 14%) but details of the phenotype are missing in reported cases.
Skeletal abnormalities
Progressive hip dysplasia and scoliosis were present in respectively 23% and 28% of the cases and could possibly be related to chronic truncal hypotonia (with or without abnormalities of limb tone), joint laxity and delayed or incomplete motor development. Surgical correction was required in five individuals in this cohort to date. The oldest individual was 18 years at time of surgery. Longer follow-up might show a larger number of surgical interventions, since the majority of included individuals (27/32) had an age of examination below 18 years. Individuals showed a tendency to short stature with five having a height ≤2.5 SD, and six others with a height between −2 and −2.5 SD. From the remaining 16 individuals, only 2 had a height >0 SD. Low bone density (z-score −4; 57%) was identified in a single case. Pes planus was present in 11 individuals.
Gastrointestinal abnormalities
Due to severe hypotonia, swallowing problems and excessive drooling were observed in more than half of the individuals (66%). Constipation was often present (62%) and may be severe, requiring the use of laxatives from early age.
Endocrine abnormalities
A diverse range of endocrine problems were reported, with low vitamin D most commonly reported (47%), and less frequently observed abnormalities including aberrant sex hormone levels, abnormal cortisol response and aberrant thyroid hormone levels (26%) (online supplementary table 1). It is likely that these problems are more common than observed, since not all individuals have been tested for endocrine abnormalities.
Congenital structural malformations
Although not frequently reported, some individuals had congenital malformations of the heart, urogenital and/or respiratory tract. Cardiac malformations included a ventricular septal defect in two individuals, and an aberrant left subclavian artery, pulmonary stenosis and mild, spontaneously closed persistent ductus arteriosus each in one individual. Abnormalities of the urogenital tract were described in five individuals: cryptorchidism in three, kidney stones in two, and congenital hydronephrosis with megaureter and urinary reflux each in one individual. Laryngeal cleft was reported in a single case.
Facial dysmorphisms
Photographs of 21 individuals are shown in figure 2. No striking similarities were observed, but a myopathic face, high anterior hairline, almond-shaped palpebral fissures and full cheeks were reported recurrently by clinicians. The presence of these features was confirmed by a panel of five independent dysmorphologists, all being asked to analyse photographs of PURA syndrome individuals from figure 2. They also mentioned eversion of lower lateral eyelids, prominent, well-defined philtrum and retrognathia as features present in a subset of individuals.

Photographs of 21 individuals with PURA mutations. Shared facial dysmorphism includes high anterior hairline, almond-shaped palpebral fissures, full cheeks and hypotonic face. Strabismus is present in several of the PURA (purine-rich element binding protein A) individuals. Additionally, independent dysmorphologists also observed eversion of lower lateral eyelids, prominent, well-defined philtrum and retrognathia in a subset of the individuals.
Expansion of the cohort with 22 previously published cases
We structurally analysed mutational and clinical data of 22 previously published individuals.6–9 An overview of published mutations is present in figure 1 and summarised clinical information is available in online supplementary table 2.
Supplementary table 2
Recurrence of mutations
We found four recurrent mutations: p.(Lys97Glu) in individual 20 and subject #4 in Lalani et al 6; p.(Phe271del) in individual 28 and subject #1 in Lalani et al 6; p(Arg245Pro) in individuals 1 and 15; p.(Phe233del) in individuals 4, 5 and 14, patient 4 in Tanaka et al 8 and patient 4 in Hunt et al.7 In addition, individuals 24 and 31 had two distinct mutations (c.153delA and c.155delG, respectively) that were both predicted to give rise to the same truncated protein product (p.(Leu54Cysfs*24)). There were six individuals between whom two distinct missense mutations were identified at each of three different amino acid positions: p.(Leu100Arg) and p.(Leu100Pro) in individual 21 and subject #1 in Lalani et al 6; p.(Ile206del) and p.(Ile206Phe) in individual 25 and patient 3 in Hunt et al 7; p.(Val226Serfs*68) and p.(Val226Glyfs*67) in individuals 6 and 10.
Clinical evaluation
Clinical features such as neurodevelopmental delay, epilepsy, and brain and ophthalmological abnormalities have been reported in all previously published cohorts. But for the majority of features, no clinical information is available across all previously published cohorts. For example, hypersomnolence, stereotypic hand movements and excessive hiccups in utero have not been reported in literature before. Using the summarised clinical information (online supplementary table 2), we calculated the frequency of clinical features in all 54 individuals (table 1).
Computational analysis of facial dysmorphism
Since no striking overlap in facial dysmorphism was observed, but similarities between several individuals have been reported by clinicians, we performed an objective computational analysis in this paper presented and previously published photographs, and compared it to the average image based on healthy controls (figure 3). Ages at photographs ranged from 2 months to 19 years. A panel of five independent dysmorphologists agreed that a myopathic face with typically open mouth appearance and full cheeks was visible on the computational modelled PURA face. Additionally, two dysmorphologists reported a slightly abnormal shape of the eyes as (1) shorter palpebral fissures and (2) eversion of lower lateral eyelids. The high anterior hairline observed in a subset of individuals (6,7,8,9,11,13,14,16,17,28) was not visible on the computational model of the PURA face.

{kind=link}
{kind=link}
{kind=link}
Computational analysis of photographs of PURA (purine-rich element binding protein A) syndrome individuals. Result of an objective computational analysis on photographs of 34 individuals with PURA mutations (ages at photographs ranging from 2 months to 19 years; right image) compared with the average image based on 301 age-matched, healthy controls (left image). The computational modelled PURA face showed a hypotonic face with typically open mouth appearance and full cheeks. Additionally, two independent dysmorphologists reported a slightly abnormal shape of the eyes as (1) shorter palpebral fissures and (2) eversion of lower lateral eyelids. The high anterior hairline observed in a subset of individuals is not visible on this computational model of the PURA face.
Modelling of all reported mutations into structural homology models of human Pur-alpha
Using the previously reported crystal structures of the N-terminal and C-terminal PUR domains from the fruit fly D. melanogaster 11 12 as templates, we generated structural homology models for the corresponding domains of the human Pur-alpha paralogue. Based on these homology-modelled structures, all 46 reported mutations were analysed and classified according to their predicted effects on domain folding and function.
The identified frameshift and non-sense mutations affecting the entire protein C-terminal to the site of mutation were defined as class A mutations. Depending on whether mutations occurred in the N-terminal PUR domain and thus affect N-terminal and C-terminal PUR domains or in the C-terminal PUR only affecting this very domain plus C-terminus, we subclassified them as A1 and A2 mutations, respectively (figure 1B). Mutations that were predicted to affect folding only locally, for instance, via amino acid exchange of a buried residue or short amino acid deletions in a folded region, were defined as class B mutations (figure 1C). Since the RNA and DNA-binding mode of Drosophila Pur-alpha has been structurally analysed,12 the human homology model could also be used to predict mutations that affect nucleic acid binding. Such mutations were found in four individuals and were defined as class C mutations (figure 1D). We also found one mutation likely affecting a surface-exposed residue that was neither predicted to impair protein folding nor nucleic acid binding. We suggest that this residue is involved in not yet understood functions such as protein–protein interactions and we defined this as class D mutation (figure 1D). Finally, we classified one mutation as class E: p.(Met1?), causing loss of the start codon. Translation is therefore likely to start at the next in-frame start codon, which is located at protein position 104. As a result, the protein will lack part of the N-terminal PUR domain, while it will have an intact C-terminal PUR domain (figure 1E). As control, we analysed variants reported by ExAC, a database containing variants of healthy controls (figure 1F). All of these variants resulted in single amino acid exchanges that are either located outside the globular domains or were exchanged against residues with similar side chain properties. Variant p.(Gln136Pro) is the only exception to this rule. It is located in the large loop region between repeats I and II and thus likely allows to compensate the changes in backbone conformation upon mutation into proline.
Genotype-phenotype analysis
Remarkable clinical variability has been observed within the total cohort of PURA syndrome individuals. To assess whether this variability could be explained by the type and/or localisation of the identified mutations, we compared (1) phenotypes of individuals with the same mutation and (2) phenotypes of individuals with a mutation of the same class.
Recurrent mutations
We observed four recurrent mutations and two mutations with a similar protein effect within the cohort, and compared their phenotypes. For all mutations, remarkable differences between individuals with the same mutation were present (table 2). For example, drug-resistant epilepsy was present in three out of five individuals with mutation p.(Phe233del) and a cardiac abnormality in two out of five individuals. The older of the two individuals without epilepsy could walk, in contrast to the three individuals with epilepsy, who were not able to walk or speak. For mutation p.(Leu54Cysfs*24), individuals had more features in common, such as epilepsy, scoliosis and short stature, but variation in ability to walk and heart abnormality was also observed.
Recurrent PURA mutations and phenotype of affected individuals
Comparison of mutation classes
We compared phenotypes of individuals subdivided by mutation class (online supplementary table 3). Overall, no significant differences in percentage of observed features between mutation classes were present. One exception is mutation p.(Glu283Argfs*45), which we classified as class D. This mutation affects only the very C-terminus (all three PUR domains are intact) and it is unlikely that this mutation interrupts protein folding or nucleic acid binding (online supplementary table 4; figure 1). Clinical features in this individual are less severe than observed in other individuals: the 14-year-old girl could walk independently from the age of 2 years and could speak in sentences. Furthermore, no neonatal problems, hypotonia, epilepsy or brain abnormalities were present.7 The clinical presentation of this girl is remarkably milder than other individuals, and could possibly be explained by the localisation of the mutation at the C-terminus.
Supplementary table 3
Supplementary table 4
Discussion
We report 32 individuals with PURA syndrome, the largest cohort so far. We systematically collected mutation and clinical data on these and previously published individuals. With information on a total of 54 individuals, we were able to delineate the clinical spectrum of this recently discovered syndrome.
The detailed and systematic collection of phenotypic data allowed us to observe less frequently reported features related to PURA syndrome. Clinical problems such as excessive hiccups in utero (55% in our cohort), hypersomnolence (66% in our cohort), stereotypic hand movements (36% in our cohort), regression since onset of seizures (38% of individuals with epilepsy in our cohort), soft skin (46% in our cohort) and short stature (19% in our cohort) have not been reported before in published studies. Post-term birth (56% in our cohort), gastro-oesophageal reflux disease (28% in our cohort), hypothermia (37% in our cohort), hypotonia after the neonatal period (97% in our cohort), constipation (62% in our cohort) and endocrine abnormalities (47% in our cohort) only have been mentioned in one other study. Similar to many other recently discovered novel syndromes for which only a small subset of individuals have been described initially, frequencies of features related to PURA syndrome were difficult to estimate based on reported numbers. By expanding the cohort, we were able to report a significant number of features not formally associated with PURA syndrome before. These features could be helpful for diagnosis and management of PURA syndrome individuals.
The phenotype of PURA syndrome has been reported as difficult to recognise in daily clinical practice. Here, we show with the identification of one individual using targeted Sanger sequencing in a neonate, that it is possible to recognise PURA syndrome based on clinical features. Severe neonatal problems including hypotonia, respiratory and feeding difficulties, hypersomnolence and hypothermia, in the majority combined with post-term birth, could point towards the early diagnosis of PURA syndrome. Although not a consistent feature, the presence of myoclonic jerks in a neonate with other features of PURA syndrome may be a useful feature to distinguish from Prader-Willi syndrome or peripheral neuromuscular disorders such as spinal muscular atrophy (SMA). Known syndromes such as congenital hypoventilation syndrome [MIM209880], Prader-Willi syndrome [MIM176270], Rett syndrome, Angelman syndrome [MIM160900], myotonic dystrophy [MIM160900] and SMA [MIM253300] have been tested often in PURA syndrome individuals. We recommend to considerPURA syndrome in the differential diagnosis of any child tested for one or more of the above syndromes, especially in the context of the aforementioned neonatal problems and/or unexplained developmental delay.
After diagnosis of PURA syndrome, careful management of existing medical problems and evaluation of health issues associated with PURA syndrome is essential. Our phenotypic overview shows that different organ systems could be affected. Therefore, we recommend routine care in a multidisciplinary team, with at least a (child) neurologist, ophthalmologist, orthopaedic surgeon and paediatrician for children aged below 18 years involved. Epilepsy, present in half of the cohort, is a major problem. Some individuals have never been seizure free and regression of achieved developmental milestones since onset of seizures has been reported. In future, studies focusing on epilepsy will be important to optimise treatment of seizures related to PURA syndrome.
In general, individuals with epilepsy were more severely affected than individuals without epilepsy. However, this explained only partially the variation in developmental phenotypes observed between individuals. To assess whether the clinical variability could be related to the type and localisation of the identified mutations, we performed a genotype-phenotype study by analysing recurrent mutations and classifying mutations based on a homology model derived from a crystal structure of the Drosophila PUR-alpha homologue. Interestingly, remarkable differences, such as the presence or absence of epilepsy or congenital malformations, were observed between individuals with the same mutation. For the remaining mutations, class D mutations with localisation at the very end of the C-terminus could possibly be correlated with a less severe phenotype. But for the other mutations, phenotypic variability and severity could not be related to the localisation and type of mutation. Based on these results, we suggest that other genetic and biological mechanisms contribute to the phenotypic variability. Further studies are needed to obtain insight into these underlying pathological mechanisms.
With the identification of 54 individuals within 2.5 years of the initial description of the condition, PURA syndrome appears to be a relatively frequent cause of ID. Previously, frequencies between 0.3% and 0.5% in neurodevelopmental cohorts have been reported.6–8 Within our cohort, 10 individuals were identified in the diagnostic lab facility of the Radboudumc, Nijmegen. These were found in a total of 4700 individuals (~0.2%) with ID referred for WES. This frequency is probably an underestimate. It has been reported that PURA, a single-exon gene rich in guanine-cytosine content, could be challenging to capture, similar to the first exon of multiexonic genes.23 Furthermore, all 10 mutations have been identified after the introduction of PURA as an ID gene in diagnostic gene panels at the end of 2014, while WES had already been in use in the diagnostic lab of the Radboudumc from 2013. Therefore, to more accurately gauge the frequency within this population, a targeted re-evaluation of all available exome data would be necessary.
In conclusion, we present a detailed overview of the clinical spectrum of PURA syndrome and a computational modelled PURA face, which is essential for better recognition of this newly recognised disorder and surveillance and management of symptoms after initial diagnosis. Our suggested approach for genotype-phenotype analysis by modelling of identified PURA mutations into human homology models of experimentally determined crystal structures from Drosophila Pur-alpha, showed that clinical variation observed between individuals is probably not related to type and localisation of mutations, but should be sought in other genetic and biological mechanisms.
Acknowledgments
We would like to thank the PURA Syndrome Foundation and the families of those individuals with PURA syndrome for their support and participation. We thank Jane Hurst, Ruth Newbury-Ecob and Karen Temple for their expert opinion on dysmorphology of PURA Syndrome. We thank Alex Paciorkowski for useful discussions. The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
References
Footnotes
DH and DB contributed equally.
Contributors All authors contributed significantly to this research and in the preparation of the manuscript. MRFR is the first author and along with DH and DB (senior authors) coordinated the group. RJ and DN performed structural modelling of mutations and contributed to the genotype-phenotype analysis. MA and CN performed computational modelling of the PURA face. JES, TvE, MV, RPWR, SJCS, APAS, JS, RP, KvD, ES, CTRMS, LAB, JMC, ME, SM, MW, KEC, SD, NSC, ECT, RF, AHML, JR, AG, TL, PI, SW, IR, KSC, JJD, DLL, KS, PL, JN, JLW, SK, JCS, SFS, HGB, CVH, MA, VEC, VMS, DDD, PS and RJL collected mutational and phenotypic data and contributed to interpretation of the data. All authors have been involved in the drafting, critical revision and final approval of the manuscript for publication. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding DH and DB are supported by a grant from the Newlife Foundation, UK. RJL is supported by a Melbourne Children’s Clinician Scientist Fellowship. DN is supported by the Deutsche Forschungsgemeinschaft NI 1110/4-1. KS and PL (JN) are supported by MH CR AZV 15-33041A. The work for patient 29 was partially funded by NMRC/CG/006/2013 from the National Medical Research Council, Ministry of Health, Republic of Singapore. The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (grant number HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (grant number WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. None of the funders were involved in study design, data collection, analysis and interpretation, writing of the report or in the decision to submit the paper for publication.
Competing interests None declared.
Patient consent Guardian consent obtained.
Ethics approval Medical Ethics Committee of the Radboud University Medical Center, Nijmegen. The study has UK Research Ethics Committee approval granted by the Cambridge South REC (10/H0305/83) and by the Republic of Ireland REC (GEN/284/12).
Provenance and peer review Not commissioned; externally peer reviewed.